 Date: April 20th & 23rd, 2015.
Date: April 20th & 23rd, 2015.exceRpt small RNA data analysis pipeline Demo
Date: April 20th & 23rd, 2015.
Presenters
Sai Lakshmi Subramanian (Primary Contact)
William Thistlethwaite
Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX
Data Coordination Component (DCC) of exRNA Communication Consortium
Robert Kitchen
Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT
Data Integration and Analysis Component (DiAC) of exRNA Communication Consortium
DMRR Data Analysis and Bioinformatics Workshop
Date: April 17th, 2016 - Sunday Location: Bethesda, MD
Location: Bethesda, MD
Presenters
Sai Lakshmi Subramanian
William Thistlethwaite
Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX
Data Coordination Component (DCC) of exRNA Communication Consortium
Overview
To use our computational deconvolution tool, you will first need a Genboree account.
You can create your Genboree account by visiting the New User Registration page.
After creating your Genboree account, you will need to log into the Genboree Workbench to use the tool.
You can find the Workbench by going to the Genboree homepage and clicking the Workbench button at the top of the page.
Alternatively, you can bookmark the Genboree Workbench directly.
After logging into your Workbench account, you'll see a screen that looks like this:

If you look inside your group, you will see that it's currently empty:

After clicking "Refresh" at the top of the Data Selector panel (see below), you should now be able to explore your database:

We're only interested in the Files area for this tool - you can ignore Tracks, Lists & Selections, Sample Sets, and Samples.
Now that you've set up your Genboree Workbench account, the next step is to process your small RNA-seq data files through exceRpt, our small RNA-seq data processing pipeline.
After completing the tutorial, you'll want to upload your own data files to process and analyze.
For now, though, we've already uploaded a set of data files for you in the Examples and Test Data group:

The deconvolution_test_data.zip archive contains 40 FASTQ files (20 plasma and 20 urine, all healthy subjects) submitted by Alessio Naccarati's group to the exRNA Atlas.
We will drag this archive to the Input Data panel on the right side of the Workbench.
Then, we will drag the database that we created earlier to the Output Targets panel.
The output files from exceRpt will be uploaded to this database.

Next, we'll select exceRpt from the tool menu at the top of the Workbench:

exceRpt has many different options (see our tutorial for more information!), but we're only going to change three of them for this submission.
First, we will update the Analysis Name so that it includes some additional information about our submission.
The analysis name will be used to organize the output files from your submission.
We recommend that you always keep a timestamp of some kind in your analysis name, as it'll help you remember when you submitted each analysis.
Second, we will update 3' Adapter Sequence from "Auto-detect 3' adapter" to "MANUALLY SPECIFY 3' ADAPTER".
Then, in the Manual Input of 3' Adapter Sequence option that appears, we will put AGATCGGAAGAGCACACGTCT.
We are providing the 3' adapter sequence manually (as opposed to having exceRpt guess the sequence) because these samples have a 3' adapter sequence which is not in exceRpt's standard adapter sequence library.

Third, we will enable the Suppress Individual Sample Emails option. Normally, you will receive one email for each sample that is processed - since we are submitting 40 samples, we don't want to receive 40 emails!
This option will suppress these individual sample emails, but you will still receive a few other emails informing you about the progress of your submission.
The option can be found under the Other Advanced Options menu at the bottom of the tool dialog - you will need to expand the menu by clicking the *+*.

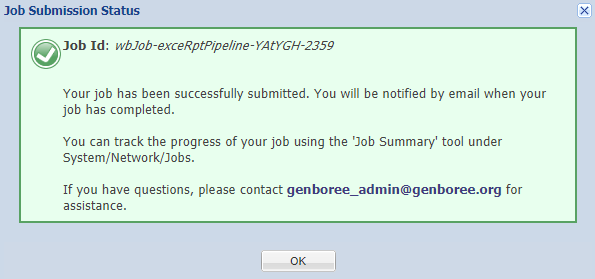
After changing these three settings, we'll click Submit.
You should see a notification informing you that your samples have been submitted:
Before proceeding to part 3 of the tutorial, you'll need to wait for your samples to be processed.
Depending on how busy our cluster is, this could take several hours.
If you don't want to wait, you can access the same results via the Examples and Test Data group:

We'll describe how to find and/or create both files below.
After your samples have been processed, you'll want to find the results created by exceRpt.
You'll find those results in your database organized by the analysis name you provided:

If you're interested in learning more about your results, you can read our data analysis tutorial.
However, for the deconvolution tool, we're really only interested in one file:

This archive contains RPM-normalized read counts for all of the different ncRNA species mapped by exceRpt (miRNA / piRNA / tRNA / GENCODE annotations / circular RNA).
These read counts are the input data for the deconvolution tool.
Your file will have a slightly different name than mine because your analysis name is different.
The second file required by the deconvolution tool is a text file that contains metadata describing the samples.
You can find an example of this metadata text file in the Examples and Test Data group:

When you're working with your own samples, you'll create your own metadata file describing your samples and upload it to your database.
IMPORTANTLY, the sample names provided in your metadata file must match the output generated by exceRpt.
During processing, exceRpt will transform the names of your FASTQ files by inserting "sample_" at the beginning and substituting underscores ("_") for any periods, pipes ("|"), or spaces.
To verify that you are providing the correct sample names in your metadata file, you can download the _exceRpt_miRNA_ReadsPerMillion.txt file generated by exceRpt and double-check that the sample names in your metadata file match what is provided there:

For the tutorial, you can just drag our pre-made file into the Input Data panel.
Finally, make sure that you dragged your database to the Output Targets panel.
Your Workbench should now look something like this:

To run the deconvolution tool, select exRNA Computational Deconvolution from the tool menu at the top of the Workbench:

This tool is much simpler than exceRpt - just provide an updated analysis name (much like you did when launching your exceRpt analysis) and click the Submit button.
The tool will likely only take a few minutes to run. Upon completion, you will receive an email informing you that your analysis is ready.
Your deconvolution results will be uploaded to your database organized by the analysis name you provided:

You can select any of the output files (explained in more detail below) and then click the "Click to Download File" link in the Details panel to download the output file.
In particular, we recommend downloading the _deconvolutionResults archive, as it will contain all results generated by the tool.
Output from the tool includes:
You can ignore the jobFile.json file. This file just contains various internal settings used to process your submission.
Overview
This tool will test samples for differential expression using DESeq2 (version 1.6.3).
Currently, the tool allows you to test a given factor (disease, for example) across two different factor levels (control versus Alzheimer's disease, for example).
We will continue to develop this tool and will add new features (like allowing analysis over multiple factors) in the coming months.
What is a  Group? Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
What is a  Database? Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each database can be associated with a reference genome. |
This step is required.
Make sure that you pick the proper reference sequence genome (hg19, for example) when creating your database.
Note that we do not have an entry for hg38 or mm10 in our "Reference Sequence" list.
If your data is associated with either of these reference sequence genomes, follow the directions below.
Create a Database for hg38 or mm10
In order to create a database associated with hg38 or mm10, select the "User Will Upload" option for "Reference Sequence"
and provide appropriate values for the Species and Version text boxes as given below:
| Your Genome of Interest | Species | Version |
| Human genome hg38 | Homo sapiens | hg38 |
| Mouse genome mm10 | Mus musculus | mm10 |
If your genome of interest is not available, please contact the exRNA Team for help.
What types of files can be uploaded?
The Fold Change Calculating Using DESeq2 tool accepts exactly two text files as input.
One file should contain your miRNA read counts, with rows corresponding to miRNA identifiers and columns corresponding to individual sample names.
The other file should contain your sample descriptors, with rows corresponding to individual sample names and columns corresponding to factor names ("condition", "biofluid", etc.).
Input Data panel. You can also drag a folder or file entity list if it contains both text files.Database to the Output Targets panel to store results.Transcriptome » Differential Expression Analysis » Fold Change Calculating Using DESeq2 from the Toolset menu.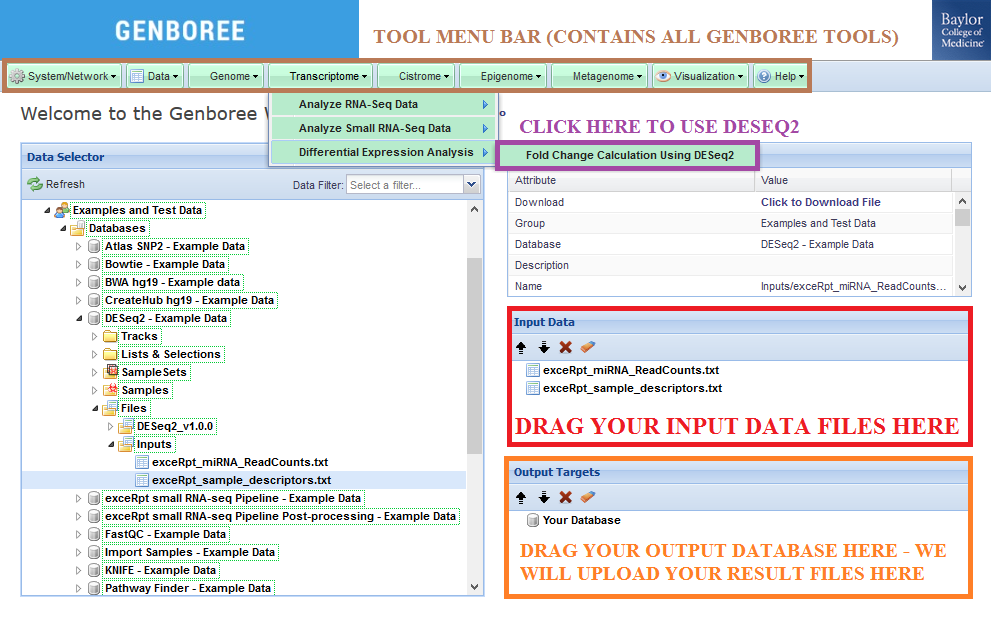
Database. The results data will end up under the DESeq_v1.0.0 folder in the Files area of your output database.
Analysis Name will be used as a sub-folder to hold the files generated by that run of the tool.Click to Download File from the Details panel to download that output file.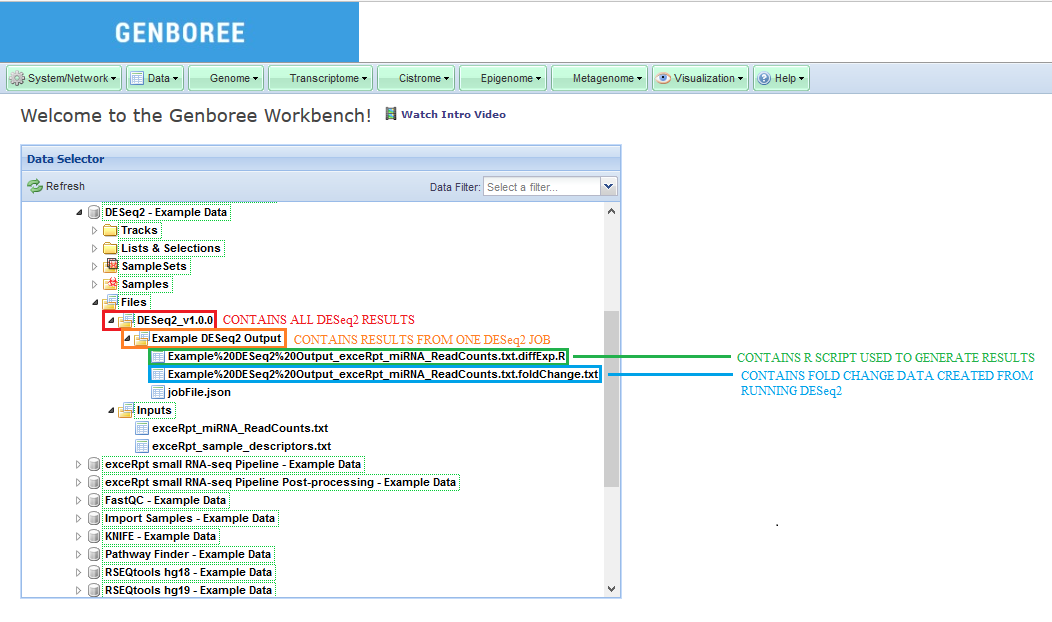
In this example, we have used a set of miRNA read counts processed by exceRpt for 181 different samples (found in exceRpt_miRNA_ReadCounts.txt).
We have also used a sample descriptor document which contains information about disease and biofluid for each of the 181 samples (found in exceRpt_sample_descriptors.txt).
The sample input files and output results can be found here:
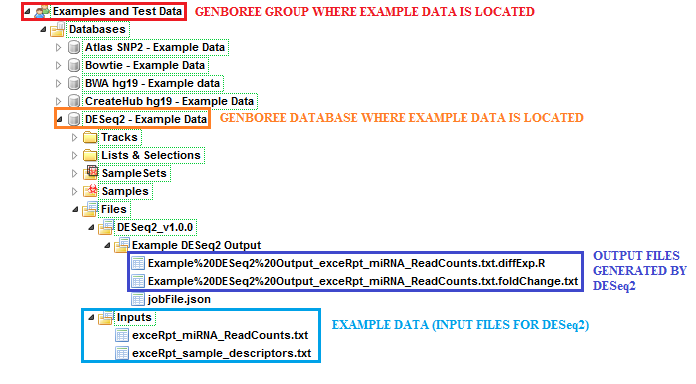
Examples and Test Data, select the database DESeq2 - Example Data.Files » Inputs.Files » DESeq2_v1.0.0 » Example DESeq2 Output folder in this database.After your job successfully completes, you will be able to download 2 different output files:
This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact the exRNA Team with questions or comments, or for help using it on your own data.
Overview
Much of the material on this page has been taken from the exceRpt GitHub page.
Many of the files generated by exceRpt for a given sample will include that sample's name.
We use the term [sampleName] to refer to this name in a general sense.
The first place to look when trying to understand your results is the .stats file.
Your .stats file can be found directly inside the directory associated with your exceRpt run on Genboree.
It can also be found in the CORE_RESULTS archive generated from your exceRpt run (further described below).
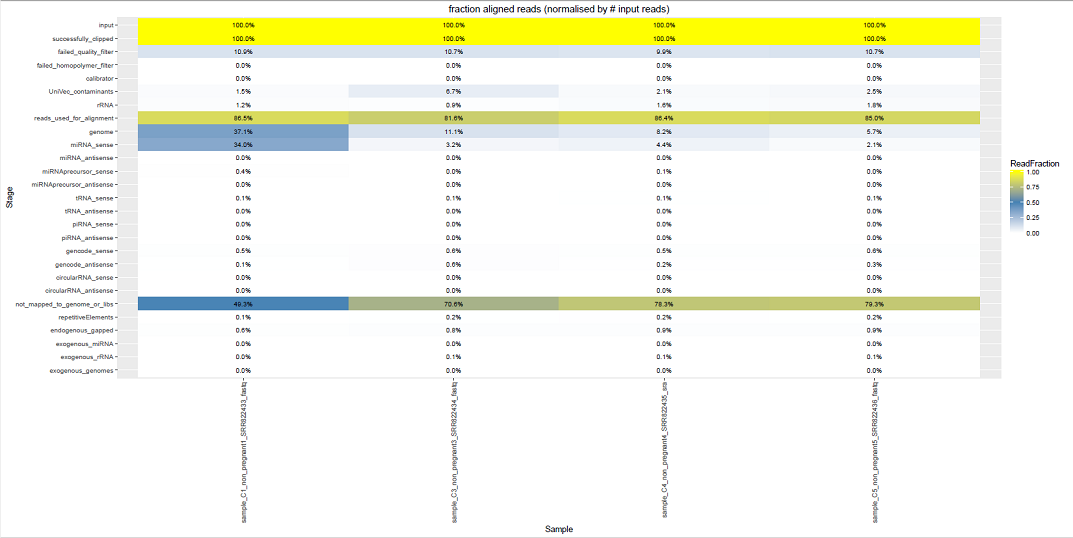
This file summarizes the number of reads that map to each class of targets in the pipeline.
In order to better understand how the pipeline maps to each class of targets (endogenous miRNAs, tRNAs, exogenous miRNAs, etc.), click here.
This link contains a useful infographic and an explanation of how the pipeline works.
Here is an example output for a .stats file:
#STATS from the exceRpt smallRNA-seq pipeline v.4.3.2 for sample sample_C5_non_pregnant5_SRR822437_fastq. Run started at 2016-07-12--12:03:16 Stage ReadCount input 1291553 successfully_clipped 1291498 failed_quality_filter 138130 failed_homopolymer_filter 49 calibrator NA UniVec_contaminants 32636 rRNA 23020 reads_used_for_alignment 1097663 genome 72996 miRNA_sense 26683 miRNA_antisense 0 miRNAprecursor_sense 386 miRNAprecursor_antisense 1 tRNA_sense 1685 tRNA_antisense 0 piRNA_sense 0 piRNA_antisense 0 gencode_sense 7189 gencode_antisense 3555 circularRNA_sense 0 circularRNA_antisense 0 not_mapped_to_genome_or_libs 1024667 #END OF STATS from the exceRpt smallRNA-seq pipeline. Run completed at 2016-07-12--15:15:46
The .stats file above was generated by an exceRpt run with exogenous mapping disabled (endogenous-only).
Your .stats file will have more information if you choose a different exogenous mapping setting.
If you choose the "endogenous + exogenous (miRNA)" setting, your .stats file will have the following extra lines:
repetitiveElements 2080 endogenous_gapped 11627 input_to_exogenous_miRNA 1010951 exogenous_miRNA 9 input_to_exogenous_rRNA 1010942 exogenous_rRNA 852
Finally, if you choose the most extensive exogenous option, "endogenous + exogenous (miRNA + Genome)", your .stats file will also have these lines:
input_to_exogenous_genomes 1010090 exogenous_genomes 2807
You can learn more about the different exogenous settings by viewing the tutorial on exceRpt's settings.
The next place to look for your analysis is the CORE_RESULTS archive uploaded with every successful exceRpt run.
This archive will be sufficient for most analyses.
We decompress the archive for you in the Genboree Workbench - you can find its contents in the CORE_RESULTS sub-folder associated with a particular run.
When you decompress your CORE_RESULTS archive (or look at the contents on Genboree), you will immediately see the following files in the base directory:
| File Name | Description of File |
| [sampleName].log | Text file containing logging information for this run |
| [sampleName].qcResult | Text file containing a variety of QC metrics for this sample |
| [sampleName].stats | Text file containing a variety of alignment statistics for this sample |
In general, you shouldn't need to look at the .log file. It contains a detailed log of the different steps performed during the course of the pipeline.
We are happy to look at the .log file to help you if something goes wrong with exceRpt or if you have a question for us.
The .qcResult file will contain data for the QC metrics discussed here.
In addition, the transcriptome complexity provided in the .qcResult file is calculated by dividing the total number of unique sequence alignments by the total number of sequence alignments when aligning to the transcriptome.
The alignments used in this calculation are taken from the endogenousAlignments_Accepted.txt.gz file (described in more detail below and only available in the full results archive).
The .stats file was discussed above.
There will be a folder in your CORE_RESULTS archive that matches the name of your sample. That folder will contain the following files:
| File Name | Description of File |
| readCounts_*_sense.txt | Read counts of each annotated RNA using sense alignments |
| readCounts_*_antisense.txt | Read counts of each annotated RNA using antisense alignments |
| *.coverage.txt | Contains read-depth across all gencode transcripts |
| *.CIGARstats.txt | Summary of the alignment characteristics for genome-mapped reads |
| [sampleName].*_fastqc.zip | FastQC output both before and after UniVec/rRNA contaminant removal |
| [sampleName].*.readLengths.txt | Counts of the number of reads of each length following adapter removal |
| [sampleName].*.counts | Read counts mapped to UniVec & rRNA (and calibrator oligo, if used) sequences |
| [sampleName].*.knownAdapterSeq | 3' adapter sequence guessed (from known adapters) in a given sample |
| [sampleName].*.adapterSeq | 3' adapter used to clip the reads in a given run |
| [sampleName].*.qualityEncoding | PHRED encoding guessed for the input sequence reads |
If you chose the "endogenous + exogenous (miRNA)" setting (mapping to exogenous miRNA / rRNA),
there will be an additional subfolder named EXOGENOUS_miRNA which will include some additional readCounts files
for exogenous miRNA. There are no readCounts files for exogenous rRNA.
Finally, if you chose the "endogenous + exogenous (miRNA + Genome)" setting (mapping to all of the above as well as exogenous genomes),
there will be an additional subfolder named EXOGENOUS_genomes which will include a taxonomy tree file
named ExogenousGenomicAlignments.result.taxaAnnotated.txt.
This text file will provide taxonomy information about the different taxons found in your sample.
When looking at the files above, you'll probably be most interested in the readCounts files.
An example of how these files are formatted can be seen below:
| ReferenceID | uniqueReadCount | totalReadCount | multimapAdjustedReadCount | multimapAdjustedBarcodeCount |
| hsa-miR-143-3p:MIMAT0000435:Homo:sapiens:miR-143-3p | 1235 | 4147219 | 4147219.0 | 0.0 |
| hsa-miR-10b-5p:MIMAT0000254:Homo:sapiens:miR-10b-5p | 1430 | 2420500 | 2420241.0 | 0.0 |
| hsa-miR-10a-5p:MIMAT0000253:Homo:sapiens:miR-10a-5p | 1115 | 784863 | 784600.5 | 0.0 |
| hsa-miR-192-5p:MIMAT0000222:Homo:sapiens:miR-192-5p | 759 | 559068 | 558542.5 | 0.0 |
Below, you can see a description of each column:
If your exceRpt run didn't map to a given library, there will be no corresponding readCounts file in your CORE_RESULTS archive.
For example, if you didn't have any tRNA sense reads, there will be no [sampleName].readCounts_tRNA_sense.txt file.
If the files given above are not sufficient, you can select the "Upload Full Results" option when launching your exceRpt job.
This will make your exceRpt job upload an archive containing all files created by exceRpt during the processing of your sample(s).
This means that your full results archive will contain all of the files located in your CORE_RESULTS archive.
Because some of the files inside this archive can be large, it is not recommended to choose this option unless you absolutely need these files.
When you open this archive, you will see a folder with your sample name (just like the CORE_RESULTS archive).
Inside that folder, you will see the following types of files:
Intermediate files containing reads 'surviving' each stage
In order of the exceRpt workflow, these files include the reads remaining after:The names of these files will look like the following:
| File Name | Description of File |
| [sampleName].*.fastq.gz | Reads remaining after each QC / filtering / alignment step |
The one exception is the read file associated with reads remaining after exogenous rRNA alignment.
This file ends in .fq.gz.
Reads aligned at each step of the pipeline
In order of the exceRpt workflow, these files include reads aligned at the following stages:The names of these files will look like the following:
| File Name | Description of File |
| filteringAlignments_*.bam | Alignments to the UniVec and rRNA sequences |
| endogenousAlignments_genome*.bam | Alignments (ungapped) to the endogenous genome |
| endogenousAlignments_genomeMapped_transcriptome*.bam | Transcriptome alignments (ungapped) of reads mapped to the genome |
| endogenousAlignments_genomeUnmapped_transcriptome*.bam | Transcriptome alignments (ungapped) of reads not mapped to the genome |
Alignment summary information obtained after invoking the library priority
By default, the library priority will choose a miRBase alignment over any other alignment.
For example, if a read is aligned to both a miRNA in miRBase and a miRNA in Gencode, the miRBase alignment is kept and all others discarded.
It is especially important for tRNAs to be chosen in favour of piRNAs, as the latter have quite a large number of misannotations compared to the former.
The names of these files will look like the following:
| File Name | Description of File |
| endogenousAlignments_Accepted.txt.gz | All compatible alignments against the transcriptome after invoking the library priority |
| endogenousAlignments_Accepted.dict | Contains the ID(s) of the RNA annotations indexed in the fifth column of the .txt.gz file above |
If you submitted dozens or even hundreds of samples for processing, you might not want to crawl through each sample's read count files.
In this situation, we recommend looking at your submission's post-processed results.
This tool combines the information from each sample into comprehensive files that cover all of the samples.
For example, if you submitted 100 samples for processing, you could look at the [analysisName]_miRNA_ReadCounts.txt file
in your post-processed results to see miRNA read counts for all 100 samples at the same time.
You can learn more about these results here.
Overview
This tool performs statistically based splicing detection for circular and linear isoforms from RNA-Seq data.
The tool's statistical algorithm increases the sensitivity and specificity of circularRNA detection from RNA-Seq data by quantifying circular and linear RNA splicing events at both annotated and un-annotated exon boundaries.
| What is a Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
| What is a Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each database can be associated with a reference genome. |
This step is required.
Make sure that you pick the proper reference sequence genome (hg19, for example) when creating your database.
The KNIFE tool currently supports the following genomes: hg19, mm10, rn5, and dm3.
However, we do not have entries for mm10, rn5, or dm3 in the reference sequence list within our Create Database tool.
If you want to create a Database associated with mm10, rn5, or dm3, select the "User Will Upload" option for "Reference Sequence".
Then, provide appropriate values for the Species and Version text boxes, as given below:
| Your Genome of Interest | Species | Version |
| Mouse genome mm10 | Mus musculus | mm10 |
| Rat genome rn5 | Rattus norvegicus | rn5 |
| Fly genome dm3 | Drosophila melanogaster | dm3 |
All data (inputs and outputs) associated with a given genome should go into that Database.
For example, if you create an hg19 Database, then all of your hg19 data belongs in that Database.
If you have other data that corresponds to another reference genome (mm10, for example), then you should create a second Database to hold that data.
What types of files can be uploaded?
The KNIFE Workbench tool accepts any number of single-end or paired-end FASTQ files as input for a single submission.Your inputs can be compressed in one large archive, multiple archives, etc.
All that matters for processing is that your paired-end FASTQ file names (not the archives containing the FASTQ files!) must follow the naming convention above.
Single-end files should either NOT include a suffix or should end in _1 or _R1.
Input Data panel. The input file(s) can be compressed. Input Data panel. Database to the Output Targets panel to store results.Transcriptome » Analyze Small RNA-Seq Data » exRNA Data Analysis » KNIFE (Known and Novel IsoForm Explorer) from the Toolset menu.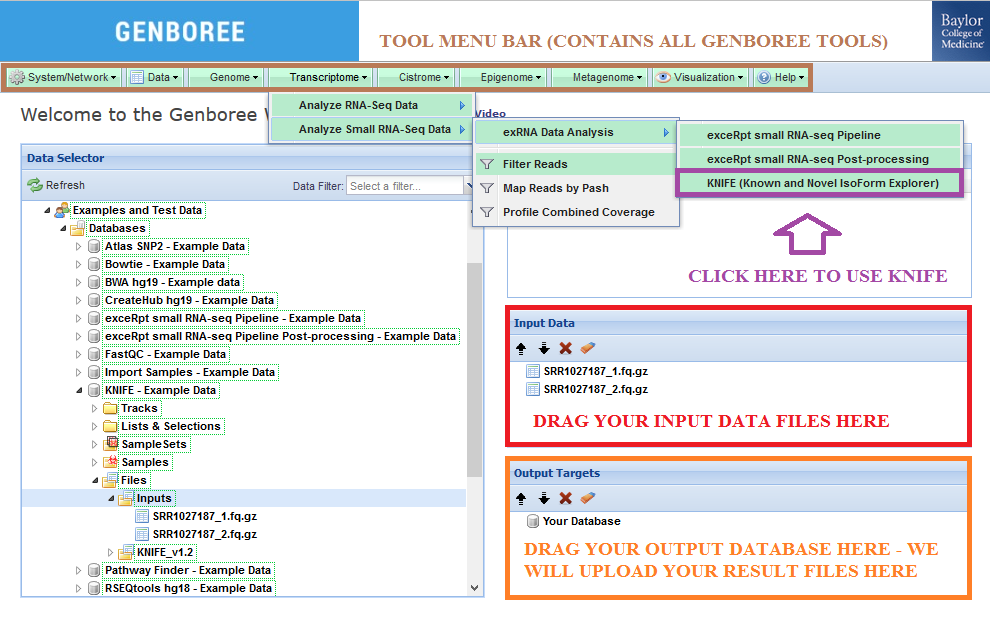
Database. The results data will end up under the KNIFE_v1.2 folder in the Files area of your output database.
Analysis Name will be used as a sub-folder to hold the files generated by that run of the tool.Click to Download File from the Details panel to download that output file.
RECOMMENDATION:
IMPORTANT NOTES:
C:/Users/John/Desktop/Submission, I would use the "cd" command in my terminal and type cd C:/Users/John/Desktop/Submission
zip command with the -X parameter (to avoid saving extra file attributes) to compress your files.johnSubmission.zip
zip -X johnSubmission.zip inputSequence1.fq.gz inputSequence2.fq.bz2 inputSequence3.fq.zip inputSequence4.sra
In this example, we have used a single sample with paired end read data from a human sample. Raw reads were grabbed from SRA, and
TrimGalore (wrapper for cutadapt) was used to trim poor quality ends and the adapter sequence. The original reads were 60nt - after
trimming, we kept all reads where both mates were at least 50nt. The FASTQ files use phred64 encoding (which is required).
The sample input FASTQ files and output results can be found here:
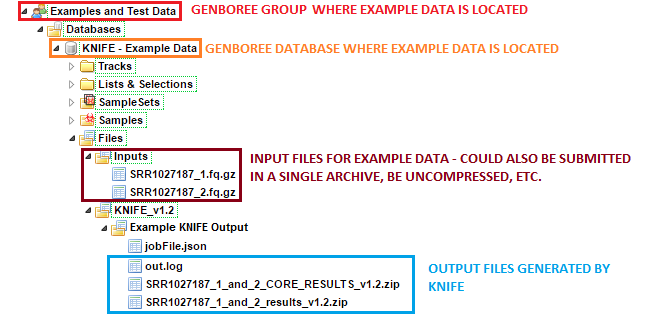
Examples and Test Data, select the database KNIFE - Example Data.Files » Inputs.Files » KNIFE_v1.2 » Example KNIFE Output folder in this database.
SRR1027187_1_and_2 subfolder contains the result files generated for the SRR1027187_1 and SRR1027187_2 paired end reads.After your job successfully completes, you will be able to download 3 different output files:
Within the full results archive, you will find four different subdirectories located in /outputs/[sample name]. These subdirectories are detailed in full below:
The core results archive will contain all of the files above except for the orig directory (which contains all sam/bam file output and is very large).
This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact Emily LaPlante with questions or comments, or for help using it on your own data.
Overview
 View Screencast (no audio)
View Screencast (no audio)
 FAQ. This step is optional. You can also use your default/existing group.
FAQ. This step is optional. You can also use your default/existing group.
| What is a Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
FAQ. This step is optional. You can also use your default/existing database.
| What is a Database? |
A Database contains Tracks, Lists, Sample Sets, Samples, and Files. Each database can be associated with a reference genome. |
FAQ. This step is REQUIRED.
What is a  Redmine Project? Redmine Project? |
The Redmine Project holds files (HTML, plots, etc) that contain analysis results from your tool. |
FAQ
| What type of files can be uploaded? | The long RNA-Seq pipeline using RSEQtools accepts a single-end or paired-end FASTQ files as input. The input files can be compressed. |
Input Data panel. The input files can be compressed. Database and a Project to Output Targets panel to store results.Transcriptome » Analyze RNA-Seq Data » Analyze RNA-Seq data by RSEQtools from the Toolset menu.Database. The results data will end up under the RSEQtools folder in the Files area of your output database.Analysis Name will be used as a sub-folder to hold the files generated by that run of the tool.
Data Selector panel.Click to Download File from the Details panel to download your results file(s).Projects page.
Data Selector panel. Link to Project in the Details panel to view your Projects page.Database to Output Targets panel.Data » Databases » Unlock/Lock Database from the Toolset menu.Output Targets panel.Database to Input Data panel. Visualization » UCSC Genome Browser from the Toolset menu.A sample from a deep-sequencing study to analyze the transcriptome changes that occur during the
differentiation of human embryonic stem cells into the neural lineage has been used in this example.
The sample consists of 27 nucleotide single-end reads, that are aligned to human reference genome build hg18
and to a splice junction library generated from the UCSC Known Genes annotation set using Bowtie2.
The mapped reads are then analyzed using various modules in RSEQtools.
Sample datasets with input and output files can be found here:
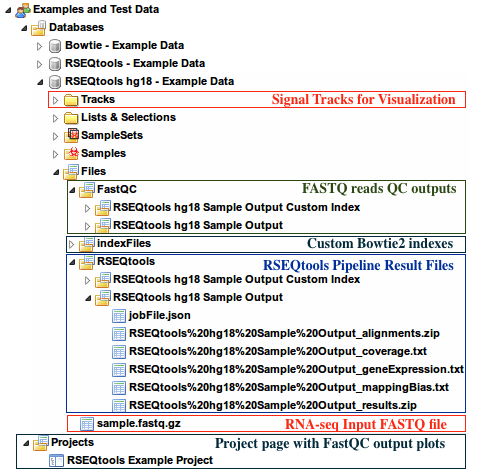
Examples and Test Data, select the database RSEQtools hg18 - Example DataFiles » sample.fastq.gzFiles » RSEQtools folder of this databaseProjects pageFiles » indexFiles » bowtie » [Your custom index folder]Tracks section of this database.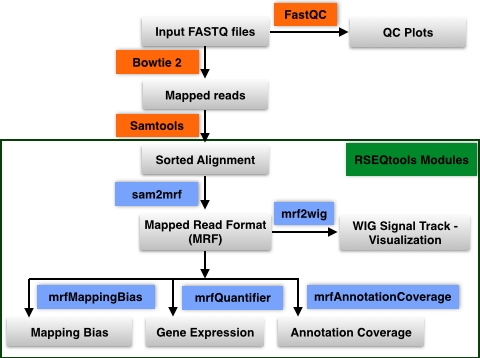
This module calculates expression values (RPKM; read coverage normalized per million mapped nucleotides
and the length of the annotation model per kb). Given a set of mapped reads in MRF and an annotation set
(representing exons, transcripts, or gene models) mrfQuantifier calculates an expression value for each annotation entry.
This is done by counting all the nucleotides from the reads that overlap with a given annotation entry.
Subsequently, this value is normalized per million mapped nucleotides and the length of the annotation item per kb.
Module to calculate mapping bias for a given annotation set. Aggregates mapped reads that overlap with
transcripts (specified in file.annotation) and outputs the counts over a standardized transcript
(divided into 100 equally sized bins) where 0 represents the 5' end of the transcript and
1 denotes the 3' end of the transcripts. This analysis is done in a strand specific way.
Module to calculate annotation coverage. Sample a set of mapped reads and determine the
fraction of transcripts (specified in annotation file) that have at least -times uniform coverage.
Generates signal track (WIG) of mapped reads from a MRF file. By default, the values in the
WIG file are normalized by the total number of mapped reads per million.
Only positions with non-zero values are reported.
Live demos of small and long RNA-Seq pipeline given at the break-out sessions.
Date: Monday, May 19th, 2014. Time: 10.35 a.m. and 3.15 p.m. Location: Conf Room C
Time: 10.35 a.m. and 3.15 p.m. Location: Conf Room C
Presenters
Sai Lakshmi Subramanian – sailakss@bcm.edu (Primary Contact)
Kevin Riehle – riehle@bcm.edu
Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX
Data Coordination Component (DCC) of exRNA Communication Consortium
Robert Kitchen - rob.kitchen@yale.edu
Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT
Data Integration and Analysis Component (DiAC) of exRNA Communication Consortium
You can download a copy of the presentation in PDF format from here:  Presentation
Presentation
Small RNA-Seq Data Analysis Tools in the Genboree Workbench
Date: Thursday, May 14th, 2015. Time: 12.00 p.m. to 4.00 p.m. Location: M321/323, DeBakey Building, Baylor College of Medicine
Presenter
Sai Lakshmi Subramanian (Primary Contact)
Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX
Data Coordination Component (DCC) of exRNA Communication Consortium
ISEV Annual Meeting 2016
Dates: 4th-7th May, 2016 Location: Rotterdam, The Netherlands
Presenters
Rocco Lucero (Presenter at the Meeting)
Sai Lakshmi Subramanian
Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX
Data Coordination Component (DCC) of exRNA Communication Consortium
Live demos of DMRR Data and Metadata Submission Infrastructure given at the break-out sessions.
Date: November 6th & 7th, 2014.
Presenters
William Thistlethwaite
Aaron Baker
Kevin Riehle
Sai Lakshmi Subramanian (Primary Contact)
Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX
Data Coordination Component (DCC) of exRNA Communication Consortium
Robert Kitchen
Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT
Data Integration and Analysis Component (DiAC) of exRNA Communication Consortium
exRNA Profiling Data Submission & Analysis Infrastructure for the ERC Consortium
Date: November 9th, 2015 - Monday Location: Rockville, MD
Presenters
Sai Lakshmi Subramanian
Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX
Data Coordination Component (DCC) of exRNA Communication Consortium
Overview
We have two different tools available on Genboree for pathway and interaction analysis.
The first, Target Interaction Finder, generates miRNA-protein target interaction files for a set of miRNA identifiers,
which can be imported into downstream tools, such as Cytoscape, for network analysis and visualization.
The second, Pathway Finder, performs a search for pathways either containing miRNAs of interest
or protein targets of those miRNAs.
You can visit each tool's tutorial page below.
You can view the tutorial page for the Target Interaction Finder tool here.
You can view the tutorial page for the Pathway Finder tool here.
Overview
The Pathway Finder tool takes a column of miRNA identifiers from an input text file and performs a search for pathways either containing miRNAs of interest or protein targets of those miRNAs.
A table of pathway results and an interactive pathway viewer are displayed in an output window after the tool successfully processes the input text file.
Below, you can view an instructional video for using the Pathway Finder Tool on the Genboree Workbench:
| What is a Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
| What is a Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each database can be associated with a reference genome. |
This step is optional.
Because this tool doesn't create any on-disk output, you only need to create a Database if you want to upload your own input file(s) for processing.
If you are using an input file in a previously existing Database (like the "Pathway Finder - Example Data" Database in the "Examples and Test Data" Group),
then you do not need to create a new Database.
If you do create a Database, make sure that you pick the proper reference sequence genome (hg19, for example) when creating your database.
Note that we do not have an entry for hg38 or mm10 in our "Reference Sequence" list.
If your data is associated with either of these reference sequence genomes, follow the directions below.
Create a Database for hg38 or mm10
In order to create a database associated with hg38 or mm10, select the "User Will Upload" option for "Reference Sequence"
and provide appropriate values for the Species and Version text boxes as given below:
| Your Genome of Interest | Species | Version |
| Human genome hg38 | Homo sapiens | hg38 |
| Mouse genome mm10 | Mus musculus | mm10 |
| What types of files can be uploaded? | The Pathway Finder tool accepts a single text file as input. This text file should have a column of miRNA identifiers as its first column. |
Input Data panel.Visualization » Pathway Finder from the Toolset menu.After your input file is successfully processed, you will be able to view a table of pathway results and an interactive pathway viewer.
The first column of the table lists a clickable pathway title that updates the viewer.
The second column lists pathway identifers that link to WikiPathways.org.
The list is sorted by the number of "miRNAs" (primary) and by "miRNA Targets" (secondary) found on each pathway.
The top 20 results are listed.

This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact William Thistlethwaite with questions or comments, or for help using it on your own data.
Overview
Current version: v4.6.2 (as of 10/12/2016)
The newest version of exceRpt is 4.6.2, which contains many updates compared to the previous, 3rd generation version on Genboree (3.3.0).
We currently still give users the option to run jobs using 3rd gen. exceRpt.
Note that some images below may have slightly outdated version numbers, but the content of the images remains otherwise accurate.
To read more about recent updates to exceRpt, view the Version Updates.
| What is a Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
| What is a Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each Database can be associated with a reference genome. |
This step is required.
You can leave "Species" and "Version" blank, as those fields are not used by exceRpt.
You can also leave "Reference Sequence" as "** User Will Upload **" - exceRpt uses its own reference files, so it doesn't need to consult your Database's reference sequence.
You will receive a warning message when creating your Database - just ignore this warning and proceed.
| What types of files can be uploaded? | The exceRpt Small RNA-Seq Pipeline accepts archives containing one or more single-end FASTQ/SRA file(s) as input. Your input files for the job MUST be compressed or else the tool will reject your job. Each submitted archive can contain multiple FASTQ/SRA files, and within those archives, each FASTQ/SRA can also be compressed. |
Input Data panel. The input file(s) must be compressed. Database to the Output Targets panel to store results.Transcriptome » Analyze Small RNA-Seq Data » exRNA Data Analysis » exceRpt small RNA-seq Pipeline from the tool menu.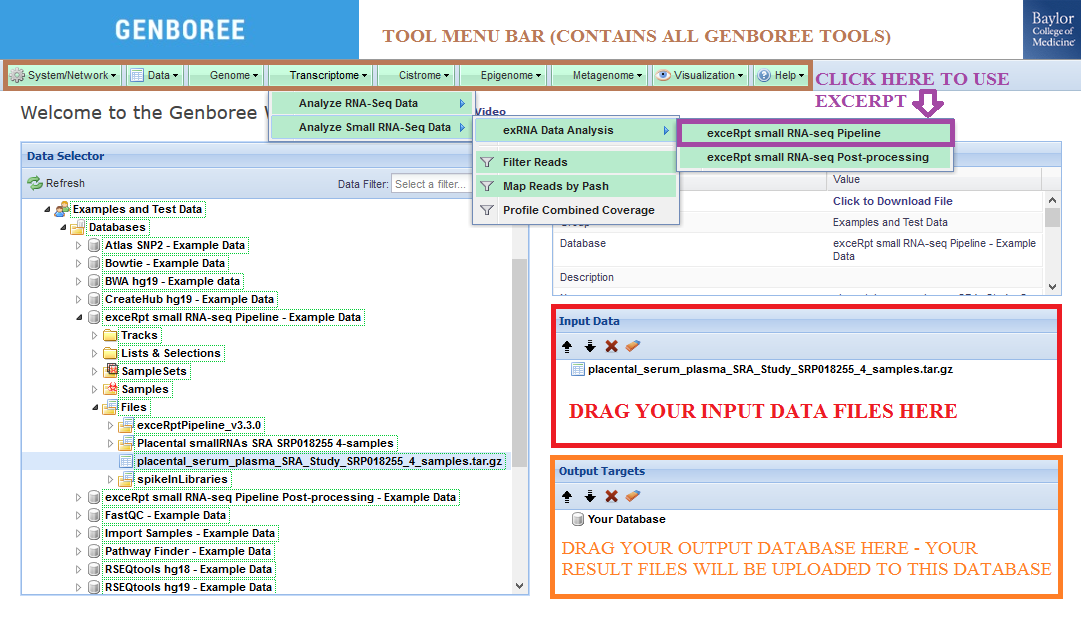
Database. The results data will end up under the exceRptPipeline_v4.6.2 folder in the Files area of your output Database.
Analysis Name will be used as a sub-folder to hold the files generated by that run of the tool.Data Selector panel.Click to Download File from the Details panel to download the results archive.CORE_RESULTS sub-folder to download the CORE_RESULTS archive (.tgz). This archive contains the most important files from your exceRpt run.
CORE_RESULTS folder on the Workbench for your convenience. Analysis Name folder, you will also find a sub-folder named postProcessedResults_v4.6.3 that contains post-processing results and plots for all of your submitted samples.

RECOMMENDATION:
IMPORTANT NOTES:
C:/Users/John/Desktop/Submission, I would use the "cd" command in my terminal and type cd C:/Users/John/Desktop/Submission
zip command with the -X parameter (to avoid saving extra file attributes) to compress your files.johnSubmission.zip
zip -X johnSubmission.zip inputSequence1.fq.gz inputSequence2.fq.bz2 inputSequence3.fq.zip inputSequence4.sra
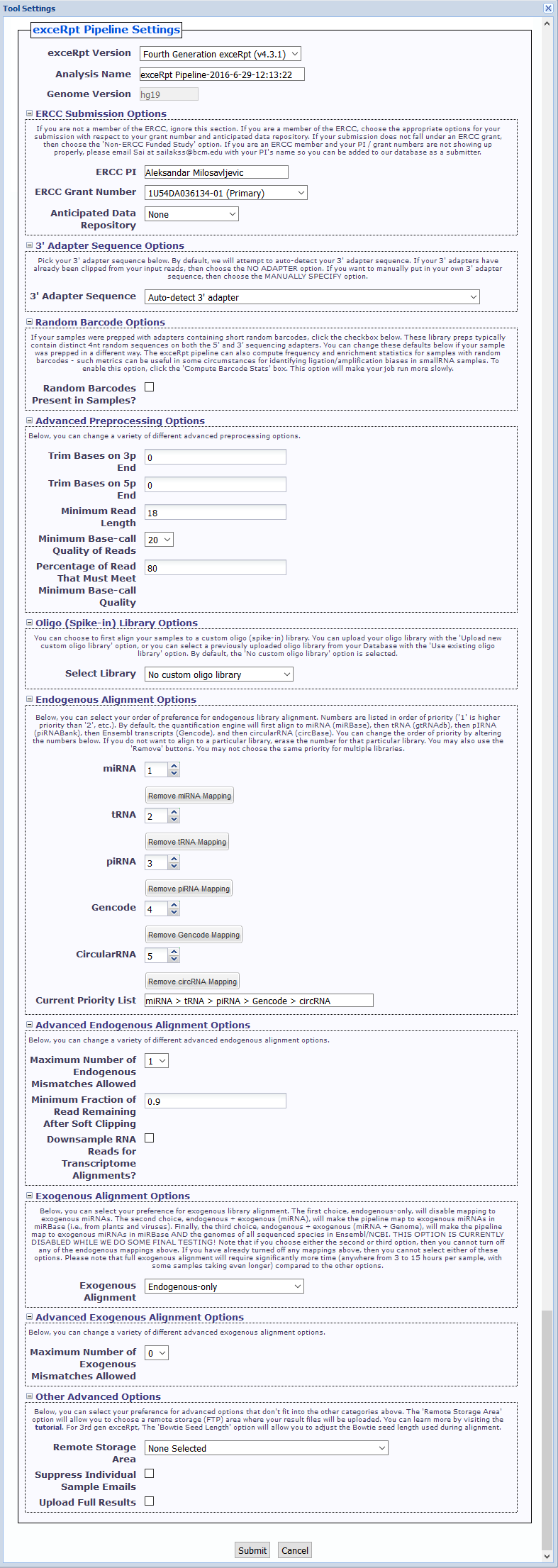
exceRptPipeline_v4.6.2) in order to organize your processed pipeline results.NO 3' ADAPTER option if your samples have already had their 3' adapters clipped.Random Barcodes Present in Samples? checkbox.Compute Barcode Stats checkbox to enable this option. Choosing this option will make your job run more slowly.Files » spikeInLibraries sub-folder in your Database for future use.In this example, we have used four samples from deep sequencing experiments of barcoded small RNA cDNA libraries to profile microRNAs in
cell-free serum and plasma from human volunteers. These samples were analyzed using the exceRpt Small RNA-seq Pipeline.
The sample input FASTQ files and output results and plots can be found here:

Examples and Test Data, select the Database exceRpt small RNA-seq Pipeline - Example Data.Files » placental_serum_plasma_SRA_Study_SRP018255_4_samples.tar.gz.
Files » Placental smallRNAs SRA SRP018255 4-samples. Files » exceRptPipeline_v4.6.2 » Circulating microRNAs from serum plasma - Study SRP18255 folder.
sample_C1_non_pregnant1_SRR822433_fastq, sample_C3_non_pregnant3_SRR822434_fastq, etc.).CORE_RESULTS folder.postProcessedResults_v4.6.3 folder (under the Circulating microRNAs from serum plasma - Study SRP18255 folder).The exceRpt Small RNA-seq Pipeline is for the processing and analysis of RNA-seq data generated to profile small-exRNAs.
The pipeline is highly modular, allowing the user to define the libraries containing smallRNA sequences that are used
during RNA-seq read-mapping, including an option to provide a library of spike-in sequences to allow absolute quantitation
of small-RNA molecules. It also performs automatic detection and removal of 3' adapter sequences.
The output data includes abundance estimates for each of the requested libraries, a variety of quality control metrics
such as read-length distribution, summaries of reads mapped to each library, and detailed mapping information for each read mapped to each library.
To better understand the results generated by exceRpt, check out the exRNA Data Analysis page.
Finally, after the pipeline finishes processing all submitted samples, a separate post-processing tool (processPipelineRuns)
is run on all successful pipeline outputs. This tool generates useful summary plots and tables that can be used to compare
and contrast different samples.
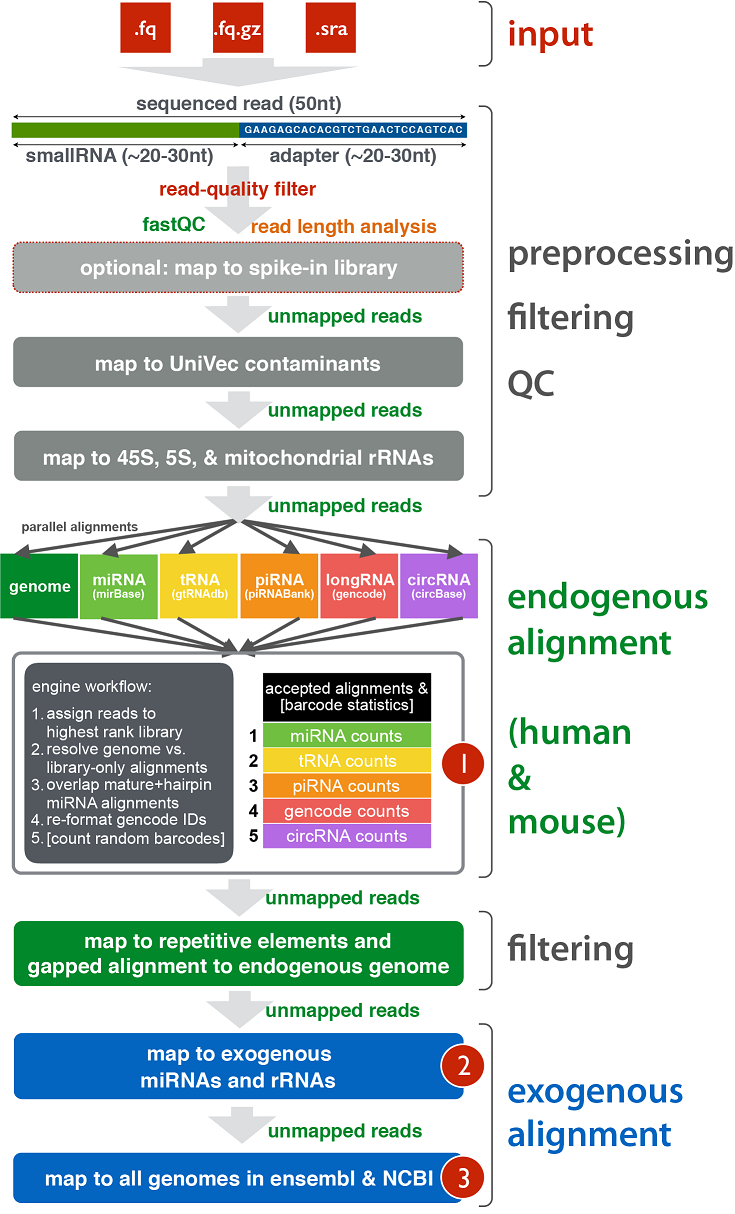

After all samples have been processed through this pipeline, the Generate Summary Report for exceRpt Results tool will take
successful samples and perform post-processing on them.
This post-processing step will generate useful plots and tables that will allow you to compare and contrast samples.
A description of all generated files can be found in the table below (partially taken from the exceRpt GitHub page):
| File Name | Description of File |
| QC Data | |
| exceRpt_DiagnosticPlots.pdf | All diagnostic plots automatically generated by the tool |
| exceRpt_readMappingSummary.txt | Read-alignment summary including total counts for each library |
| exceRpt_ReadLengths.txt | Read-lengths (after 3' adapters/barcodes are removed) |
| Raw Transcriptome Quantifications | |
| exceRpt_miRNA_ReadCounts.txt | miRNA read-counts quantifications |
| exceRpt_tRNA_ReadCounts.txt | tRNA read-counts quantifications |
| exceRpt_piRNA_ReadCounts.txt | piRNA read-counts quantifications |
| exceRpt_gencode_ReadCounts.txt | gencode read-counts quantifications |
| exceRpt_circularRNA_ReadCounts.txt | circularRNA read-counts quantifications |
| exceRpt_biotypeCounts.txt | read-counts quantified for different biotypes |
| Normalized Transcriptome Quantifications | |
| exceRpt_miRNA_ReadsPerMillion.txt | miRNA RPM quantifications |
| exceRpt_tRNA_ReadsPerMillion.txt | tRNA RPM quantifications |
| exceRpt_piRNA_ReadsPerMillion.txt | piRNA RPM quantifications |
| exceRpt_gencode_ReadsPerMillion.txt | gencode RPM quantifications |
| exceRpt_circularRNA_ReadsPerMillion.txt | circularRNA RPM quantifications |
| Exogenous Output | |
| exceRpt_exogenousGenomes_TaxonomyTrees_aggregateSamples.pdf | aggregate taxonomy tree for exogenous genomes |
| exceRpt_exogenousGenomes_TaxonomyTrees_perSample.pdf | per-sample taxonomy trees for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomyCumulative_ReadCounts.txt | descendant read counts for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomyCumulative_ReadsPerMillion.txt | descendant RPM counts for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomySpecific_ReadCounts.txt | direct read counts for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomySpecific_ReadsPerMillion.txt | direct RPM counts for exogenous genomes |
| exceRpt_exogenousRibosomal_TaxonomyTrees_aggregateSamples.pdf | aggregate taxonomy tree for exogenous rRNAs |
| exceRpt_exogenousRibosomal_TaxonomyTrees_perSample.pdf | per-sample taxonomy trees for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomyCumulative_ReadCounts.txt | descendant read counts for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomyCumulative_ReadsPerMillion.txt | descendant RPM counts for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomySpecific_ReadCounts.txt | direct read counts for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomySpecific_ReadsPerMillion.txt | direct RPM counts for exogenous rRNAs |
| R Objects | |
| exceRpt_smallRNAQuants_ReadCounts.RData | All raw data (binary R object) |
| exceRpt_smallRNAQuants_ReadsPerMillion.RData | All normalized data (binary R object) |
| Misc. | |
| exceRpt_adapterSequences.txt | 3' adapter sequences associated with each sample |
| exceRpt_sampleGroupDefinitions.txt | sample group associated with each sample (not used in Genboree) |
The taxonomy specific read counts file contains the number of reads that map directly to each node in the sample's taxonomy tree.
The taxonomy cumulative read counts file contains the number of reads that map to the descendants of each node in the sample's taxonomy tree.
Importantly, the reads counted for a given node in the taxonomy specific read counts file are NOT counted as part of the cumulative counts in the other file
(all of the node's descendants are counted, but not the node itself).
This means that no reads mapped directly to the Viruses node, but a considerable number of reads mapped to descendants of Viruses.
Similarly, say we have this line in the cumulative file:This means that a considerable number of reads mapped both directly to the cellular organisms node as well as descendants of that node.
In other words, if you want to get a full count of all of the reads that aligned to a given node and its descendants,
you need to ADD TOGETHER the numbers in both the taxonomy cumulative and taxonomy specific files for that node.
You can also find plots of the exogenous trees in the .pdf plot files.
First, the TaxonomyTrees_perSample.pdf file contains a plot for each sample.
The percentage within each node is the ratio of the node's summed cumulative + specific reads to the root node's summed cumulative + specific reads.
Second, the TaxonomyTrees_aggregateSamples.pdf file contains a single plot that condenses all samples into a single, averaged tree.
The percentage within each node is the ratio of that node's summed cumulative + specific reads averaged across all samples to the root node's summed cumulative + specific reads averaged across all samples.
Below, we can see what the post-processing files look like on the Genboree Workbench (found in the Examples and Test Data Group):

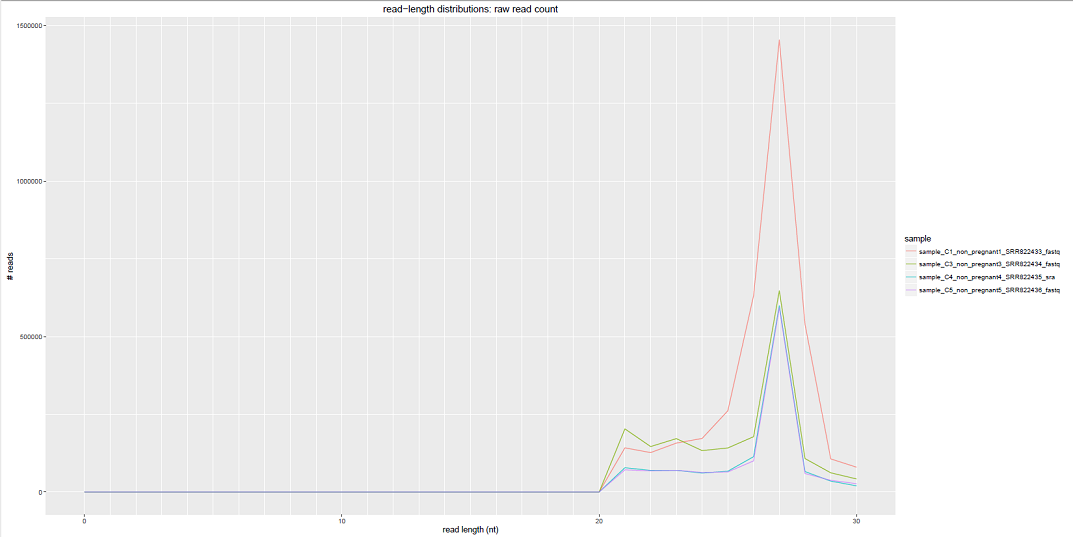
We can see some examples below of the comparative plots available in the DiagnosticPlots.pdf generated by the post-processing tool.
Each plot contains data from four different samples (the example data).


This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact "exRNA Team":brl-exrna@bcm.edu with questions or comments, or for help using it on your own data.
Overview
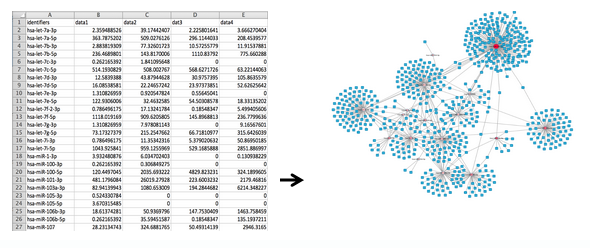
This tool takes a column of miRNA identifiers from an input TXT file and generates miRNA-protein target interaction files in both a tabular CSV format and a network XGMML format.
The target interactions are sourced from miRTarBase 1, an experimentally validated miRNA-target interaction database.
The CSV and XGMML files can be imported into downstream tools, such as Cytoscape 2, for network analysis and visualization.
From a column of identifiers... to a network of interactions
Below, you can view an instruction video for using the Target Interaction Finder Tool on the Genboree Workbench:
| What is a Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
| What is a Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each database can be associated with a reference genome. |
This step is required.
Make sure that you pick the proper reference sequence genome (hg19, for example) when creating your database.
Note that we do not have an entry for hg38 or mm10 in our "Reference Sequence" list.
If your data is associated with either of these reference sequence genomes, follow the directions below.
Create a Database for hg38 or mm10
In order to create a database associated with hg38 or mm10, select the "User Will Upload" option for "Reference Sequence"
and provide appropriate values for the Species and Version text boxes as given below:
| Your Genome of Interest | Species | Version |
| Human genome hg38 | Homo sapiens | hg38 |
| Mouse genome mm10 | Mus musculus | mm10 |
| What types of files can be uploaded? | The Target Interaction Finder tool accepts one or more text files as input. Each text file should have a column of miRNA identifiers as its first column. Your input text files can also be compressed, with each archive containing containing one or more input text files. For example, you could submit one archive containing 100 different input text files, or even three different archives containing different numbers of input text files. |
Input Data panel. These input file(s) can also be compressed, as mentioned above.Database to the Output Targets panel to store results.Visualization » Target Interaction Finder from the Toolset menu.Database. The results data will end up under the targetInteractionFinder folder in the Files area of your output database.
Analysis Name will be used as a sub-folder to hold the files generated by that run of the tool.Click to Download File from the Details panel to download that output file.After your job successfully completes, you will be able to download 3 different output files:
This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact William Thistlethwaite with questions or comments, or for help using it on your own data.
Overview
We have recently implemented some storage quotas for using exceRpt.
In particular, these quotas apply when you select the "Upload Full Results" option.
Please read the information below to better understand the storage quotas
and how you can clear up space for your Genboree Group.
Each Genboree Group has two different storage quotas associated with it.
These storage quotas include local space and FTP-backed space.
Note that the quotas are associated with a Group and not a user!
Please do not create multiple Groups as a single user to bypass the quota restriction.
You should only use a single Group for your exceRpt output (unless discussed with us previously).
By default, a Genboree Group is allotted 100 GB of local space and 100 GB of FTP-backed space.
Local space is defined as any space outside of the virtual FTP areas in your Group.
You can learn more about creating virtual FTP areas on the Workbench here.
As mentioned above, these space requirements only apply if you launch an exceRpt job with
the "Upload Full Results" option enabled. This option will upload a large results archive for each
sample containing alignment .bam files (and other similar files).
You can learn more about the specific files present in the full results archive on the exRNA Data Analysis page.
The default storage quotas should be sufficient for most users, especially if you normally run your samples
through exceRpt without the "Upload Full Results" option.
Any files in your Genboree Group will contribute to either the local or FTP-backed quota.
For example, if I uploaded a large number of FASTQ files that I wanted to process, those would contribute to one of the quotas.
This is one reason why it is very important to compress your files on the Workbench.
You can learn more about this idea below.
When you submit your files for processing through exceRpt (with "uploadFullResults" enabled),
the following factors will be considered:
1) How much space you're currently taking up in your Group (discussed above)
2) An estimation of how much space your current submission will take up when fully uploaded
3) An estimation of how much space your other, currently-running submissions will take up when fully uploaded
In other words, if I launch a huge job with 200 samples, I will likely get an error message when I try to launch another job,
even if my first job hasn't finished yet.
If you do receive an error message, it will clearly indicate how much space is taken up by each of the enumerated factors above.
Because we only recently instituted these quotas, many Genboree Groups have far exceeded the numbers given above
and cannot launch exceRpt jobs until space is freed up.
We have a number of suggestions for meeting the quotas:
If you have any files that you no longer need on the Workbench, you can delete those files to free up space.
You should use the Remove File(s) tool (found under Data -> Files) to do so.
Simply drag your files into the Input Data panel and then run the tool to delete them.
The tool also accepts folders as inputs, so if you want to delete an entire folder at once, you can do that also.
Remember that exceRpt is constantly being worked on and improved, so if you have some old results, you could
potentially delete those and re-run your samples through the newest version of exceRpt.
Another way to make space is to compress your files if they are currently uncompressed.
For example, many users have uncompressed FASTQ files stored on Genboree.
These FASTQ files can be very large, but they get much smaller when they're compressed.
Furthermore, we recently added a restriction to exceRpt so that it no longer accepts uncompressed FASTQ files as input.
This means that if you want to use FASTQ files in exceRpt, you will need to compress them first.
You can compress the files on the Genboree Workbench using the Prepare Archive tool (found under Data -> Files).
Simply drag your uncompressed files into the Input Data panel and the Database where you want your archive created
into the Output Targets panel.
IMPORTANT: After you run the tool and receive an email informing you that the job was successful,
you must manually delete your old uncompressed files. We will implement an option in the near future
that will do this deletion automatically, but for now, you'll need to do it using the Remove File(s) tool (discussed above).
As mentioned above, we have two different quotas: one for local storage and one for FTP-backed storage.
Thus, if one quota is completely full, it might make sense to move some files to a different storage type to free up space.
You can accomplish this by using the "Copy / Move File" tool (found under Data -> Files).
In most cases, users will want to move their files from local storage to FTP-backed storage, so we'll explain how to do that below.
If you do have files in your FTP-backed area that you want to move to local storage, the method is very similar.
Drag the files you want to move into the Input Data panel, and then drag the Database where you want your files to be stored
into the Output Targets panel.
Before requesting more space, you should try all of the methods given above.
If you still need more space, email Emily and let him know your Genboree Group and why you need more space.
For example, if your Group stores files associated with a collaborative effort between several different labs,
then it might make sense to increase that Group's storage quota.
Below, we'll go step-by-step through the process of setting up your remote storage area on Genboree and then downloading your exceRpt result files.
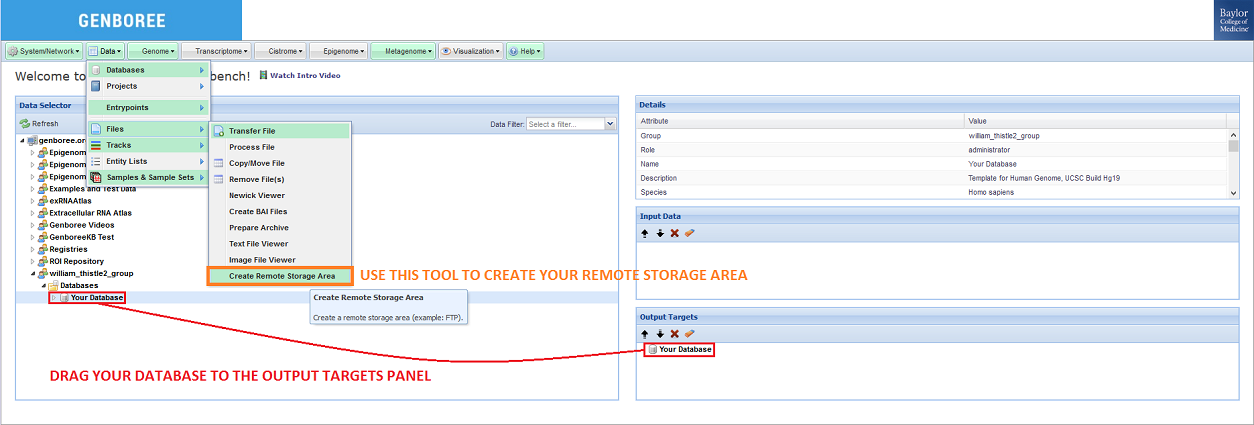
The first step in the process is creating a remote storage area in your Database of choice.
If you're unfamiliar with Genboree, you should first learn about Groups and then learn about creating Databases.
Note that your account comes with a default group named after your user login.
After you have created a Database, you should drag it from the Data Selector panel to the Output Targets panel.
Then, you can select the Create Remote Storage Area tool in the tool menu at the top of the screen:
When you click the Create Remote Storage Area button, a window like the following will appear:
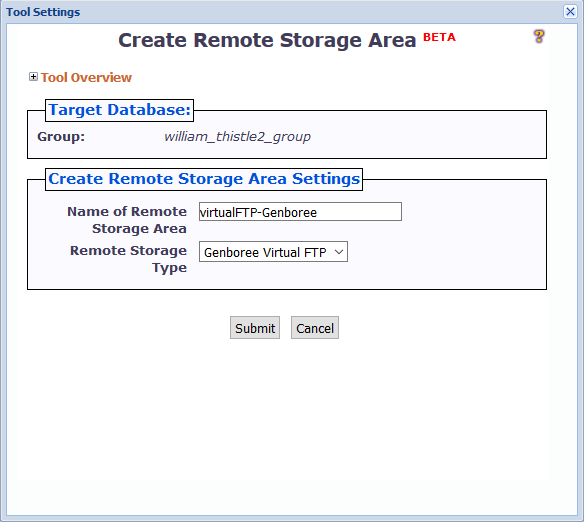
There are two different settings you can change:
You can name the remote storage area anything you want, so long as the name is unique among folders in the Files area of your Database.
This is because the remote storage area is represented as a folder in the Files area.
Under "Remote Storage Type", you can select the particular type of remote storage area that you want to create.
Currently, we only support the Genboree FTP server - thus, you can go ahead and keep the default option of "Genboree Virtual FTP".
After you click "Submit", you will receive notification that your remote storage area was successfully created (or an informative error message if something went wrong).
Your remote storage area will now be available in the Files area under the Database you chose:
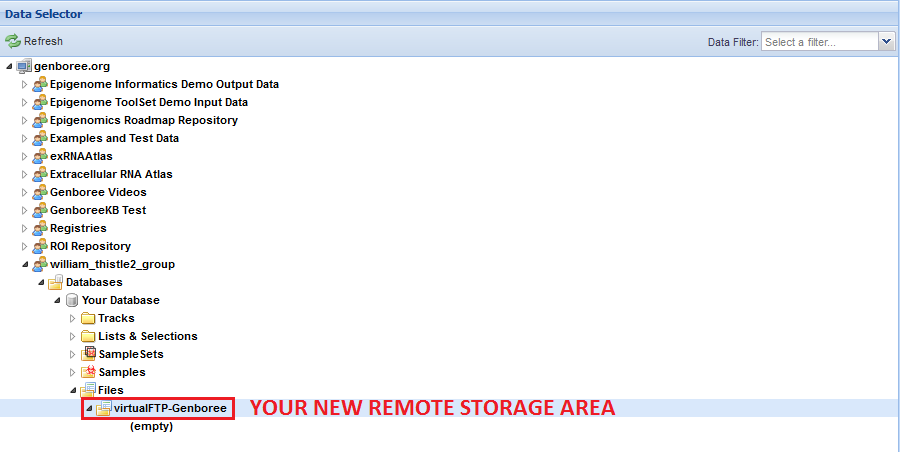
You can use this tool to create as many remote storage areas as you like, so long as they all have different names.
Now that you've created your remote storage area, the next step is to submit your files for processing through exceRpt.
This tutorial will provide guidance on submitting your samples.
In particular, you will need to select your newly created remote stage area in the Remote Storage Area menu under Advanced Options.
The default option for this menu is None Selected - you should choose the remote storage area where you want to store your exceRpt result files.
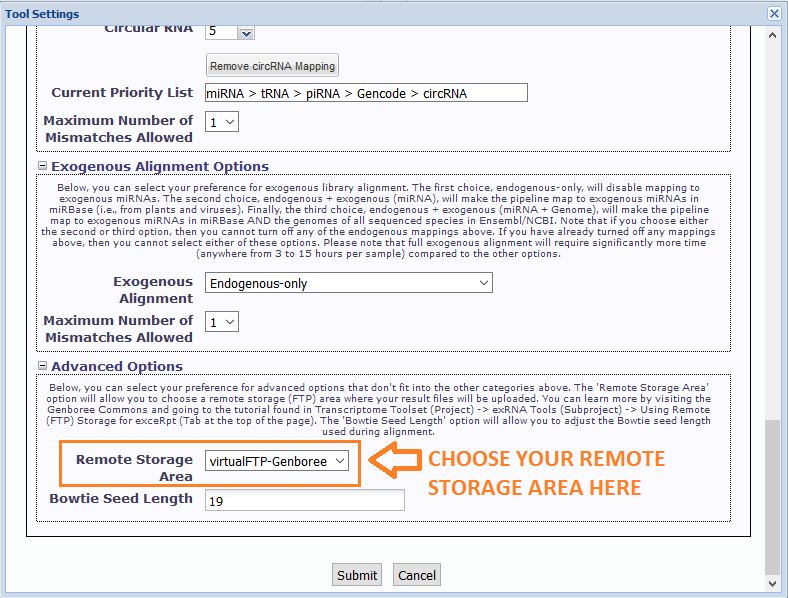
After you click "Submit", your files will be processed through exceRpt.
If any issue arises during processing, you will receive an email notifying you about the issue.
If you have additional questions about your submission, you can always email exRNA Team for help.
After we've finished processing your samples, you'll be able to access your results by both the Genboree Workbench and your FTP client.
If you want to use the Genboree Workbench, accessing your samples is just like any other exceRpt job, with one small difference.
With normal exceRpt submissions, the base directory for your exceRpt pipeline runs can be found in the Files area.
However, if you use a remote storage area, the base directory for your exceRpt pipeline runs will be located in that remote storage folder.
You can see an example below:
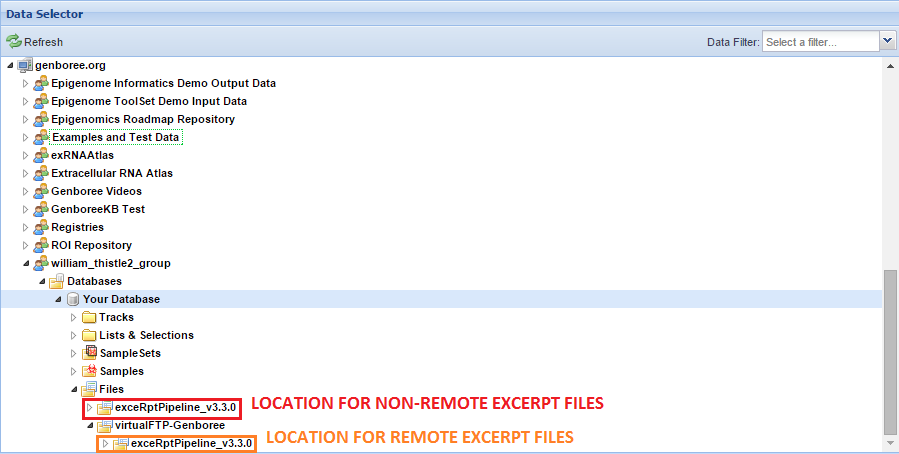
Before you can access your results, you will need an account on our FTP server.
You should email exRNA Team and ask her to create a personal FTP account for you so that you can log onto our FTP server.
Please note that creating a Genboree account does not create your FTP account as well.
You will always need to contact the exRNA Team in order to create your FTP account.
After we have confirmed that your FTP account is created, you can log into our FTP server at ftps://ftps.genboree.org with your Genboree username and password.Note: If you would like multiple people to have download permissions for this FTP folder you can include the usernames of the other people here
Please note that you must send an email to us for each new Database you want to use for remote storage.
For example, say you create a remote storage area in Database A and then ask us to expose that area to your FTP username.
We will do so, and then you will be able to see that area via your FTP client.
If you create more remote storage areas in the same Database (Database A), then you will also be able to see those areas without emailing us again.
However, if you create a new Database on Genboree (Database B) and then create a remote storage area in that Database,
you will need to email us again with the name of the new Database so we can expose it to your FTP username.
Overview
Video Tutorial - Introduction to the Genboree Workbench
FAQs Related to Genboree Workbench Basics
View Screencast (no audio)
This tool generates miRNA-protein target interaction files for a set of miRNA identifiers,
which can be imported into downstream tools, such as Cytoscape, for network analysis and visualization.
View Screencast below (no audio):
This tool performs a search for pathways either containing miRNAs of interest
or protein targets of those miRNAs.
View Screencast below (no audio):