Overview
- Detecting Circular and Linear Isoforms from RNA-seq Data Using KNIFE
- Introduction
- Preliminary Steps for Setting Up Any Analysis in the Genboree Workbench
- Create a Group for Your Analysis
- Create a Database for Your Analysis
- Upload your Data File(s)
- Step-by-step Instructions to Set Up KNIFE Submission
- Notes for Preparing Input Data Files
- Example Data for Running KNIFE
- Summary of Output Files Generated by KNIFE
- Detailed Explanation of Output Files Generated by KNIFE
- References and Attributions
Detecting Circular and Linear Isoforms from RNA-seq Data Using KNIFE¶
Introduction¶
This tool performs statistically based splicing detection for circular and linear isoforms from RNA-Seq data.
The tool's statistical algorithm increases the sensitivity and specificity of circularRNA detection from RNA-Seq data by quantifying circular and linear RNA splicing events at both annotated and un-annotated exon boundaries.
Preliminary Steps for Setting Up Any Analysis in the Genboree Workbench¶
Create a Group for Your Analysis¶

What is a  Group? Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
Create a Database for Your Analysis¶
What is a  Database? Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each database can be associated with a reference genome. |
This step is required.
Make sure that you pick the proper reference sequence genome (hg19, for example) when creating your database.
The KNIFE tool currently supports the following genomes: hg19, mm10, rn5, and dm3.
However, we do not have entries for mm10, rn5, or dm3 in the reference sequence list within our Create Database tool.
If you want to create a Database associated with mm10, rn5, or dm3, select the "User Will Upload" option for "Reference Sequence".
Then, provide appropriate values for the Species and Version text boxes, as given below:
| Your Genome of Interest | Species | Version |
| Mouse genome mm10 | Mus musculus | mm10 |
| Rat genome rn5 | Rattus norvegicus | rn5 |
| Fly genome dm3 | Drosophila melanogaster | dm3 |
All data (inputs and outputs) associated with a given genome should go into that Database.
For example, if you create an hg19 Database, then all of your hg19 data belongs in that Database.
If you have other data that corresponds to another reference genome (mm10, for example), then you should create a second Database to hold that data.
Upload your Data File(s)¶
What types of files can be uploaded?
The KNIFE Workbench tool accepts any number of single-end or paired-end FASTQ files as input for a single submission.Each single-end FASTQ file (or pair of paired-end FASTQ files) will be processed separately, with a subfolder created for each input (or pair of inputs).
Paired-end FASTQ files must follow a certain naming convention: read1 must end in _1 or _R1, and read2 must end in the accompanying suffix (_2 or _R2).
- Examples: SAMPLENAME_1.fastq and SAMPLENAME_2.fastq, SAMPLENAME_R1.fq and SAMPLENAME_R2.fq
Your inputs can be compressed in one large archive, multiple archives, etc.
All that matters for processing is that your paired-end FASTQ file names (not the archives containing the FASTQ files!) must follow the naming convention above.
Single-end files should either NOT include a suffix or should end in _1 or _R1.
Step-by-step Instructions to Set Up KNIFE Submission¶
- Drag one or more FASTQ files to the
Input Datapanel. The input file(s) can be compressed.
Optionally, you can also upload one or more compressed archives of multiple FASTQ files. Each FASTQ file can also be compressed inside these archives.
Then, you can drag these multiple input file archives to theInput Datapanel.
Check out the Notes below for more info on preparing your input data files. - Drag a
Databaseto theOutput Targetspanel to store results. - Select
Transcriptome»Analyze Small RNA-Seq Data»exRNA Data Analysis»KNIFE (Known and Novel IsoForm Explorer)from the Toolset menu.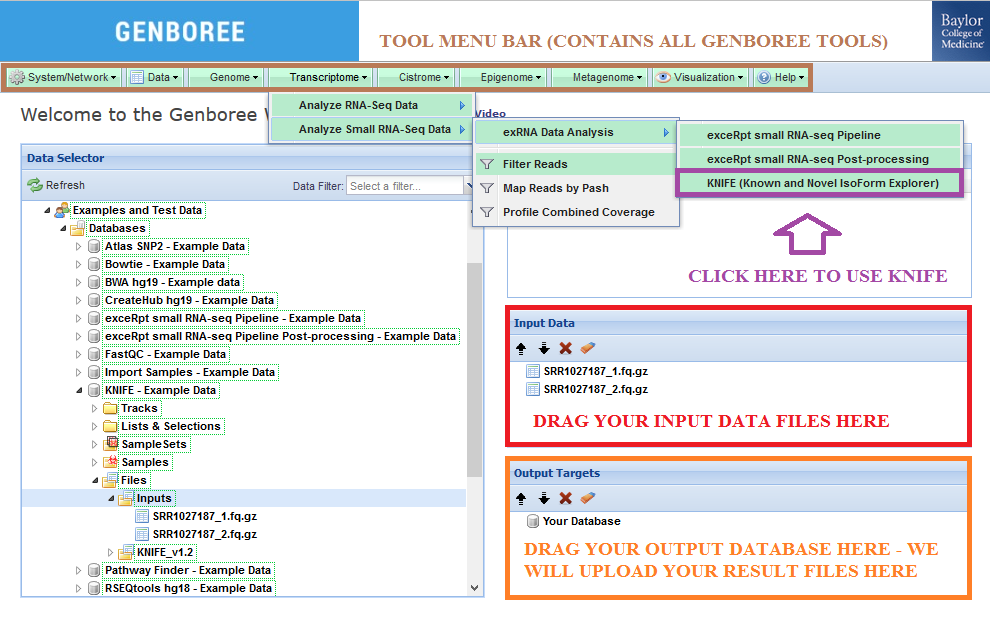
- Fill in the analysis name for your tool job. We recommend keeping a timestamp in your analysis name!
- Select the different ERCC-related submission settings if you are a member of the ERCC. If you are not, then ignore this section.
- Choose to upload your results to a remote storage area if you wish to do so. More information about this option can be found here.
- Submit your job. Upon completion of your job, you will receive an email.
- Download the results of your analysis from your
Database. The results data will end up under theKNIFE_v1.2folder in theFilesarea of your output database.- Within that folder, your
Analysis Namewill be used as a sub-folder to hold the files generated by that run of the tool. - Open this sub-folder to see your results.
- Select any of the output files (explained in more detail below) and then click the link
Click to Download Filefrom theDetailspanel to download that output file.
- Within that folder, your

Notes for Preparing Input Data Files¶
RECOMMENDATION:
- If you have a large number of input FASTQ files, it is highly recommended to make smaller archives with fewer number of input files
in order to avoid issues with uploading them to your Genboree Database.- Each compressed archive should not be larger than 10GB.
- Upload all of your smaller archives to your Genboree Database and then submit them all in the same KNIFE job.
- This technique will allow you to successfully upload many input files and compare/contrast results from those input files in one job submission.
IMPORTANT NOTES:
- If you are using Mac OS to prepare your files, remember to remove the "__MACOSX" sub-directory that gets added to the compressed archives.
- In order to create your archive using the terminal, first navigate to the directory where your files are.
- EXAMPLE: If my files were located in
C:/Users/John/Desktop/Submission, I would use the"cd"command in my terminal and typecd C:/Users/John/Desktop/Submission
- Next, you will use the
zipcommand with the-Xparameter (to avoid saving extra file attributes) to compress your files. - EXAMPLE: Imagine that I am submitting 4 data files : inputSequence1.fq.gz, inputSequence2.fq.bz2, inputSequence3.fq.zip, inputSequence4.sra
I want to name my .zip filejohnSubmission.zip- I would type the following:
zip -X johnSubmission.zip inputSequence1.fq.gz inputSequence2.fq.bz2 inputSequence3.fq.zip inputSequence4.sra
- I would type the following:
- EXAMPLE: If my files were located in
- Commonly used compression formats like .zip, .gz, .tar.gz, .bz2 are accepted.
Example Data for Running KNIFE¶
In this example, we have used a single sample with paired end read data from a human sample. Raw reads were grabbed from SRA, and
TrimGalore (wrapper for cutadapt) was used to trim poor quality ends and the adapter sequence. The original reads were 60nt - after
trimming, we kept all reads where both mates were at least 50nt. The FASTQ files use phred64 encoding (which is required).
The sample input FASTQ files and output results can be found here:
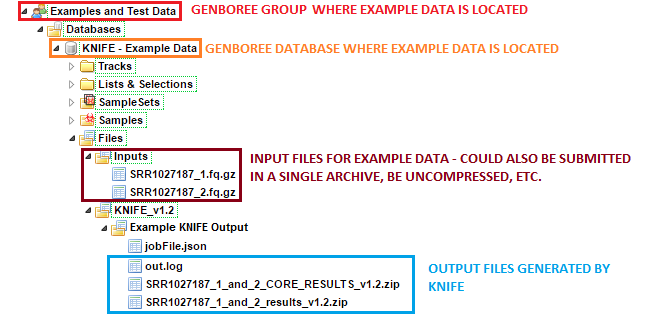
- Under the group
Examples and Test Data, select the databaseKNIFE - Example Data. - Both compressed input files can be found in the folder:
Files » Inputs. - KNIFE results can be found under the
Files » KNIFE_v1.2 » Example KNIFE Outputfolder in this database.- In particular, the
SRR1027187_1_and_2subfolder contains the result files generated for theSRR1027187_1andSRR1027187_2paired end reads. - Each processed sample is given its own dedicated folder.
- In particular, the
Summary of Output Files Generated by KNIFE¶
After your job successfully completes, you will be able to download 3 different output files:
- A _results_v1.2.zip file that contains all of the result files from your KNIFE run.
- This file will be quite large (gigabytes) because it includes all of the full alignment files for your run.
- A _CORE_RESULTS_v1.2.zip file that contains all of the result files from your KNIFE run, minus the full alignment files.
- This file is a good alternative to the full results archive, as it is significantly smaller (the archive will be ~150 megabytes).
- An out.log file that contains output generated by the KNIFE run.
Detailed Explanation of Output Files Generated by KNIFE¶
Within the full results archive, you will find four different subdirectories located in /outputs/[sample name]. These subdirectories are detailed in full below:
- circReads: The primary output files you will be interested in looking at are in the following subdirectories.
- reports: read count and p-value per junction using naive method. 2 files created per sample, 1 for annotated junctions (linear and circular) and the other for de novo junctions.
For single end reads and de novo junctions from either single end or paired end data, these are the output files of interest as GLM reports are for annotated junctions using paired end data only.
You will want to select a threshold on the p-value for which of these junctions are considered true positive circles.
For the publication, we considered all junctions with a p-value of 0.9 or higher and a decoy/circ read count ratio of 0.1 or lower.- junction: chr|gene1_symbol:splice_position|gene2_symbol:splice_position|junction_type|strand
- junction types are reg (linear), rev (circle formed from 2 or more exons), or dup (circle formed from single exon)
- linear: number of reads where read1 aligned to this linear junction and read2 was consistent with presumed splice event, or just number of aligned reads to this linear junction for SE reads
- anomaly: number of reads where read2 was inconsistent with read1 alignment to this linear junction
- unmapped: number of reads where read1 aligned to this junction and read2 did not map to any index
- circ: number of reads where read1 aligned to this circular junction and read2 was consistent with presumed splice event, or just number of aligned reads to this circular junction for SE reads
- decoy: number of reads where read2 was inconsistent with read1 alignment to this circular junction
- pvalue: naive method p-value for this junction based on all aligned reads (higher = more likely true positive).
You will want to select a threshold on the p-value for which of these junctions are considered true positive circles. - scores: (read1, read2) Bowtie2 alignment scores for each read aligning to this junction, or scores at each 10th percentile for junctions with more than 10 reads
- junction: chr|gene1_symbol:splice_position|gene2_symbol:splice_position|junction_type|strand
- glmReports: read count and posterior probability per junction using GLM (only for PE reads, annotation-dependent junctions).
2 files created per sample, 1 with circular splice junctions and the other with linear splice junctions.
You will want to select a threshold on the posterior probability for which of these junctions are considered true positive circles.
For the publication, we considered all junctions with a posterior probability of 0.9 or higher.- junction: chr|gene1_symbol:splice_position|gene2_symbol:splice_position|junction_type|strand
- junction types are reg (linear), rev (circle formed from 2 or more exons), or dup (circle formed from single exon)
- numReads: number of reads where read1 aligned to this junction and read2 was consistent with presumed splice event
- p_predicted: posterior probability that the junction is a true junction (higher = more likely true positive).
- p_value: p-value for the posterior probability to control for the effect of total junctional counts on posterior probability
- junction: chr|gene1_symbol:splice_position|gene2_symbol:splice_position|junction_type|strand
- glmModels: RData files containing the model used to generate the glmReports
- ids: alignment and category assignment per read
- reports: read count and p-value per junction using naive method. 2 files created per sample, 1 for annotated junctions (linear and circular) and the other for de novo junctions.
- sampleStats: Contains 2 txt files with high-level alignment statistics per sample (read1 and read2 reported separately).
- SampleAlignStats.txt: useful for evaluating how well the library prep worked, for example ribosomal depletion.
Number of reads are reported, with fraction of total reads listed in ()- READS: number of reads in original fastq file
- UNMAPPED: number of reads that did not align to any of the junction, genome, transcriptome, or ribosomal indices
- GENOME: number of reads aligning to the genome
- G_STRAND: percentage of GENOME reads aligning to forward strand and percentage aligning to reverse strand
- TRANSCRIPTOME: number of reads aligning to the transcriptome
- T_STRAND: percentage of TRANSCRIPTOME reads aligning to forward strand and percentage aligning to reverse strand
- JUNC: number of reads aligning to the scrambled or linear junction index and overlapping the junction by required amount
- J_STRAND: percentage of JUNC reads aligning to forward strand and percentage aligning to reverse strand
- RIBO: number of reads aligning to the ribosomal index
- R_STRAND: percentage of RIBO reads aligning to forward strand and percentage aligning to reverse strand
- 28S, 18S, 5.8S, 5SDNA, 5SrRNA: percentage of RIBO aligning to each of these ribosomal subunits (for human samples only)
- HBB: number of reads aligning to HBB genomic location (per hg19 annotation)
- SampleCircStats.txt: useful for comparing circular and linear ratios per sample
- CIRC_STRONG: number of reads that aligned to a circular junction that has a p-value >= 0.9 using the naive method (very high confidence of true circle)
- CIRC_ARTIFACT: number of reads that aligned to a circular junction that has a p-value < 0.9 using the naive method
- DECOY: number of reads where read2 did not align within the circle defined by read1 alignment
- LINEAR_STRONG: number of reads that aligned to a linear junction that has a p-value >= 0.9 using the naive method (very high confidence of true linear splicing)
- LINEAR_ARTIFACT: number of reads that aligned to a linear junction that has a p-value < 0.9 using the naive method
- ANOMALY: number of reads where read2 did not support a linear transcript that includes the read1 junction alignment
- UNMAPPED: number of reads where read1 aligned to a linear or scrambled junction but read2 did not map to any index
- TOTAL: sum of all previous columns, represents total number of reads mapped to junction but not to genome or ribosome
- CIRC_FRACTION: CIRC_STRONG / TOTAL
- LINEAR_FRACTION: LINEAR_STRONG / TOTAL
- CIRC / LINEAR: CIRC_FRACTION / LINEAR_FRACTION
- SampleAlignStats.txt: useful for evaluating how well the library prep worked, for example ribosomal depletion.
- orig: contains all sam/bam file output and information used to assign reads to categories.
In general there is no reason to dig into these files since the results, including the ids of reads that aligned to each junction, are output in report files under circReads as described above,
but sometimes it is useful to dig back through if you want to trace what happened to a particular read.- genome: sam/bam files containing Bowtie2 alignments to the genome index
- junction: sam/bam files containing Bowtie2 alignments to the scrambled junction index
- reg: sam/bam files containing Bowtie2 alignments to the linear junction index
- ribo: sam/bam files containing Bowtie2 alignments to the ribosomal index
- transcriptome: sam/bam files containing Bowtie2 alignments to the transcriptome index
- unaligned: fastq and fasta files for all reads that did not align to any index
- forDenovoIndex: fastq files containing subset of the unaligned reads that are long enough to be used for creating the denovo junction index
- denovo: sam/bam files containing Bowtie2 alignments to the de novo junction index
- still_unaligned: fastq files containing the subset of the unaligned reads that did not align to the denovo index either
- ids: text files containing the ids of reads that aligned to each index, location of alignment, and any other relevant data from the sam/bam files used in subsequent analysis.
The reads reported in the junction and reg subdirectories are only those that overlapped the junction by user-specified amount.
In juncNonGR and denovoNonGR, the reported read ids are the subset of reads that overlapped a junction and did not align to the genome or ribosomal index.
- denovo_script_out: debugging output generated during creation of de novo index.
The core results archive will contain all of the files above except for the orig directory (which contains all sam/bam file output and is very large).
References and Attributions¶
- Szabo L, Morey R, Palpant NJ, Wang PL, Afari N, Jiang C, Parast MM, Murry CE, Laurent LC, Salzman J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biology. 2015, 16:126. http://www.ncbi.nlm.nih.gov/pubmed/26076956
- Integrated into the Genboree Workbench by William Thistlethwaite and Sai Lakshmi Subramanian at the Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX.
This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact Emily LaPlante with questions or comments, or for help using it on your own data.