Overview
- Small RNA-Seq Data Analysis for exRNA Profiling Using the exceRpt Small RNA-seq Pipeline
- Version Updates
- Preliminary Steps for Setting Up Any Analysis in the Genboree Workbench
- Create a Group for Your Analysis
- Create a Database for Your Analysis
- Upload your Data File(s)
- Step-by-step Instructions for Setting Up Your exceRpt small RNA-Seq Data Analysis
- Notes for Preparing Input Data Files
- Tool Settings
- Example Data for Running exceRpt Small RNA-seq Pipeline
- exceRpt Small RNA-seq Pipeline Workflow (Implemented in the Genboree Workbench)
- exRNA Data Analysis Results
- Currently Supported Genomes
- Sources of smallRNA Libraries
- Workflow
- Example of Core Results from Workflow
- Post-processing of Samples
- Explanation of Exogenous Output
- Example of Post-processed Result Files
- Comparative Plots
- Bioinformatics Tools Used in This Pipeline (4th Gen)
- References and Attributions
Small RNA-Seq Data Analysis for exRNA Profiling Using the exceRpt Small RNA-seq Pipeline¶
Version Updates¶
Current version: v4.6.2 (as of 10/12/2016)
The newest version of exceRpt is 4.6.2, which contains many updates compared to the previous, 3rd generation version on Genboree (3.3.0).
We currently still give users the option to run jobs using 3rd gen. exceRpt.
Note that some images below may have slightly outdated version numbers, but the content of the images remains otherwise accurate.
To read more about recent updates to exceRpt, view the Version Updates.
Preliminary Steps for Setting Up Any Analysis in the Genboree Workbench¶
Create a Group for Your Analysis¶

What is a  Group? Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
Create a Database for Your Analysis¶
What is a  Database? Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each Database can be associated with a reference genome. |
This step is required.
You can leave "Species" and "Version" blank, as those fields are not used by exceRpt.
You can also leave "Reference Sequence" as "** User Will Upload **" - exceRpt uses its own reference files, so it doesn't need to consult your Database's reference sequence.
You will receive a warning message when creating your Database - just ignore this warning and proceed.
Upload your Data File(s)¶
| What types of files can be uploaded? | The exceRpt Small RNA-Seq Pipeline accepts archives containing one or more single-end FASTQ/SRA file(s) as input. Your input files for the job MUST be compressed or else the tool will reject your job. Each submitted archive can contain multiple FASTQ/SRA files, and within those archives, each FASTQ/SRA can also be compressed. |
Step-by-step Instructions for Setting Up Your exceRpt small RNA-Seq Data Analysis¶
- Drag one or more archives containing single-end FASTQ or SRA files to the
Input Datapanel. The input file(s) must be compressed.
Each FASTQ/SRA file can also be compressed inside these archives.
Check out the Notes below for more info on preparing your input data files. - Drag a
Databaseto theOutput Targetspanel to store results. - Select
Transcriptome»Analyze Small RNA-Seq Data»exRNA Data Analysis»exceRpt small RNA-seq Pipelinefrom the tool menu.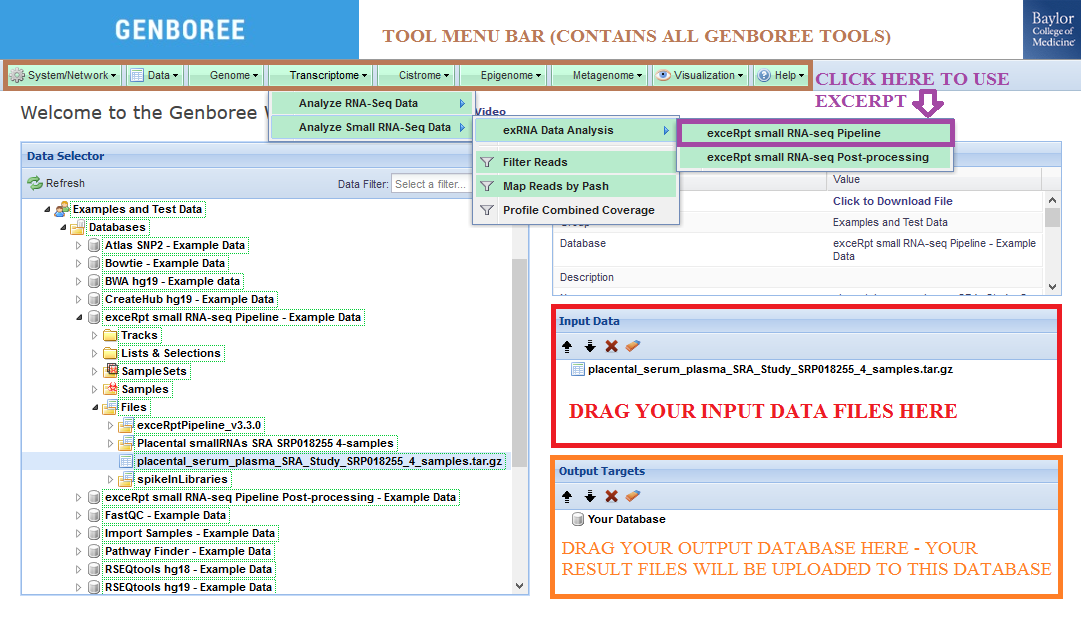
- Fill in appropriate details in the Tool Settings dialog box. See Tool Settings for more details.
- Submit your job. You will receive several emails as we process your samples.
- First, you will receive an email about your exceRpt small RNA-seq Pipeline (New, Customized Endogenous Engine) Batch Submission job.
This email just lets you know if your samples were successfully submitted for processing. - Second, you will receive one or more emails about your Run exceRpt on Single Sample job(s).
You will receive an email for each sample submitted, and those emails will let you know which samples were processed successfully.
If you selected the "Suppress Individual Sample Emails" option, you will not receive these emails. - Third, you will receive an email about your Generate Summary Report for exceRpt Results job.
This email lets you know if post-processing on your samples was performed successfully. - Fourth, you will receive an email about your ERCC Final Processing job.
This email will give you a brief summary report about the different jobs above.
- First, you will receive an email about your exceRpt small RNA-seq Pipeline (New, Customized Endogenous Engine) Batch Submission job.
- Download the results of your analysis from your
Database. The results data will end up under theexceRptPipeline_v4.6.2folder in theFilesarea of your output Database.- Within that folder, your
Analysis Namewill be used as a sub-folder to hold the files generated by that run of the tool. - In order to see the results for a particular sample, open the sub-folder corresponding to that sample.
- If you selected the "Upload Full Results" option, you will see a results .zip that will contain all result files from your exceRpt run, including any full alignment .bam files.
- Click on the results archive (.zip) in the
Data Selectorpanel. - Select the link
Click to Download Filefrom theDetailspanel to download the results archive.
- Click on the results archive (.zip) in the
- The .stats file will contain the read counts mapped at various stages of the exceRpt mapping process.
- Open the
CORE_RESULTSsub-folder to download the CORE_RESULTS archive (.tgz). This archive contains the most important files from your exceRpt run.- This archive is usually all you need from a given exceRpt run and is much smaller than the full results archive (.zip) since it doesn't contain full alignment .bam files.
- The CORE_RESULTS archive will be decompressed in the
CORE_RESULTSfolder on the Workbench for your convenience.
- Finally, under the
Analysis Namefolder, you will also find a sub-folder namedpostProcessedResults_v4.6.3that contains post-processing results and plots for all of your submitted samples.- These files are described in further detail below.
- Within that folder, your

Notes for Preparing Input Data Files¶
RECOMMENDATION:
- If you have a large number of input FASTQ/SRA files, it is highly recommended to make smaller archives with fewer numbers of input files
in order to avoid issues with uploading the archives to your Genboree Database.- Each compressed archive should be smaller than 10GB.
- Upload all of your smaller archives to your Genboree Database and then submit them all in the same exceRpt Small RNA-seq Pipeline job.
- This technique will allow you to successfully upload many input files and compare/contrast results from those input files in one job submission.
IMPORTANT NOTES:
- The archive should not contain any folders or sub-folders - all files should be directly placed into the archive.
If you are using Mac OS to prepare your files, remember to remove the "__MACOSX" sub-directory that gets added to the compressed archives. - In order to create your archive using the terminal, first navigate to the directory where your files are.
- EXAMPLE: If my files were located in
C:/Users/John/Desktop/Submission, I would use the"cd"command in my terminal and typecd C:/Users/John/Desktop/Submission
- Next, you will use the
zipcommand with the-Xparameter (to avoid saving extra file attributes) to compress your files. - EXAMPLE: Imagine that I am submitting 4 data files : inputSequence1.fq.gz, inputSequence2.fq.bz2, inputSequence3.fq.zip, inputSequence4.sra
I want to name my .zip filejohnSubmission.zip- I would type the following:
zip -X johnSubmission.zip inputSequence1.fq.gz inputSequence2.fq.bz2 inputSequence3.fq.zip inputSequence4.sra
- I would type the following:
- EXAMPLE: If my files were located in
- Commonly used compression formats like .zip, .gz, .tar.gz, .bz2 are accepted.
Tool Settings¶
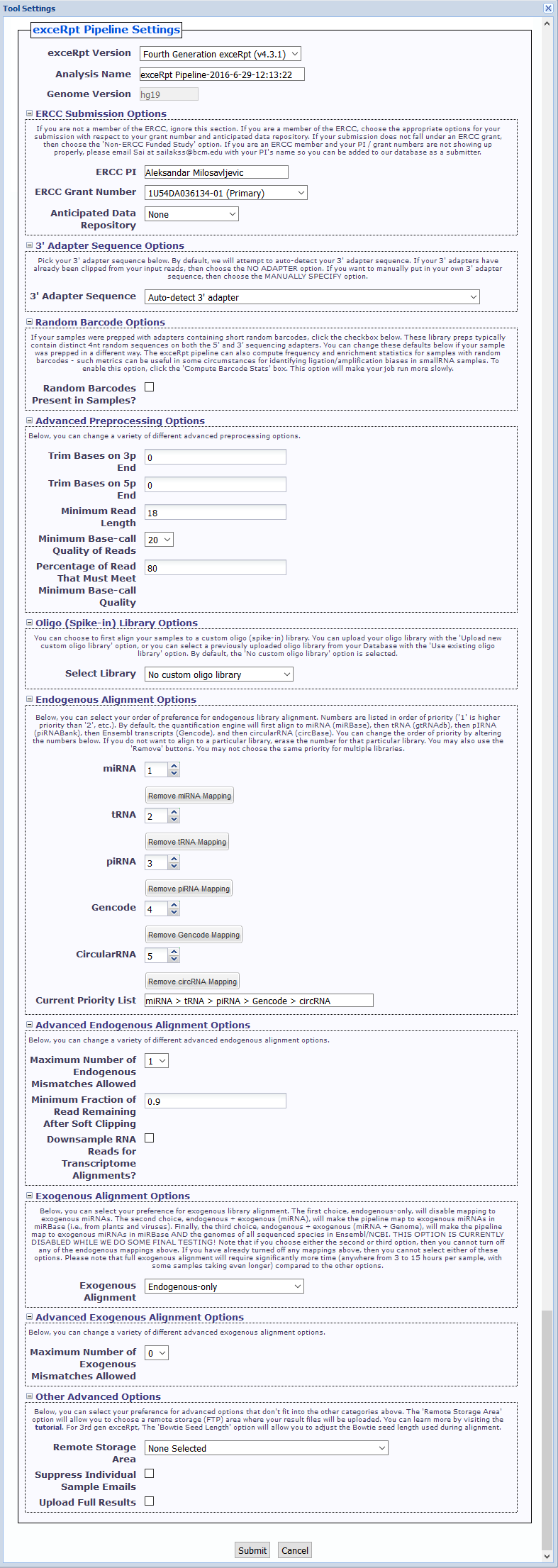
- exceRpt Version You can choose between using 4th gen exceRpt (4.6.2) and 3rd gen exceRpt (3.3.0) here.
Only settings relevant to the chosen version of exceRpt will be displayed.
We will remove the option to use 3rd gen exceRpt in the near future. - Analysis Name This name is used under the top-level output folder (
exceRptPipeline_v4.6.2) in order to organize your processed pipeline results.
You should include identifying information (timestamp, disease associated with samples, etc.) in your analysis name so you can easily distinguish between your different submissions. - Genome Version You can choose the genome version associated with your input files here. Please note that only one genome version is allowed per submission.
- ERCC Submission Options Here, if you are a member of the ERCC, you can select the grant number and anticipated data repository associated with your submission.
If your submission does not fall under an ERCC grant, then choose the 'Non-ERCC Funded Study' option.
If you are an ERCC member and your PI / grant numbers are not showing up properly, please email exRNA Team with your PI's name so you can be added to our database as a submitter. - 3' Adapter Sequence Options You can select your 3' adapter sequence here. By default, we will attempt to auto-detect the adapter sequence for your samples.
Auto-detection is a good choice if you don't know your 3' adapter sequence or if your submission includes samples with different 3' adapter sequences,
as we don't currently support manual input of multiple 3' adapter sequences in the same submission.
You can also select one of the pre-defined 3' adapter sequences, manually specify your own 3' adapter sequence, or select theNO 3' ADAPTERoption if your samples have already had their 3' adapters clipped. - Random Barcode Options If your sequences have adapter sequences that contain short random barcodes, click the
Random Barcodes Present in Samples?checkbox.
You'll then be able to enter the length and location of your random barcodes.
The exceRpt pipeline can also compute frequency and enrichment statistics for samples with random barcodes.
Such metrics can be useful in some circumstances for identifying ligation/amplification biases in smallRNA samples.
Click theCompute Barcode Statscheckbox to enable this option. Choosing this option will make your job run more slowly. - Advanced Preprocessing Options (initially minimized)
- Trim Bases on 3p End - This option will trim N bases from the 3' end of each read, where N is the value you choose. Default of 0.
- Trim Bases on 5p End - This option will trim N bases from the 5' end of each read, where N is the value you choose. Default of 0.
- Minimum Read Length - This option will alter the minimum read length we will use after adapter (and random barcode) removal. Minimum value allowed is 10. Default of 18.
- Minimum Base-call Quality of Reads - This option will alter the minimum base-call quality of reads. Default of 20.
- Percentage of Read That Must Meet Minimum Base-call Quality - This option will alter the percentage of a given read that must meet the minimum base-call quality given above. Default of 80.
- Oligo (Spike-in) Library Options - There are 3 options:
- No custom oligo library - No mapping to custom oligo library.
- Upload new custom oligo library - Upload a single FASTA file with a list of all spike-in sequences used.
Uploaded sequences are stored under theFiles»spikeInLibrariessub-folder in your Database for future use. - Use existing oligo library - Select an existing oligo library from your Database.
- Endogenous Alignment Options
- You can select your order of preference for endogenous library alignment.
- Numbers are listed in order of priority ('1' is higher priority than '2', etc.).
- By default, the quantification engine will first align to miRNA, then tRNA, then pIRNA, then Gencode annotations, and then circular RNA.
- You can change the order of priority by altering the numbers next to each library.
- If you do not want to align to a particular library, erase the number for that particular library. You may also use the "Remove" buttons.
- You may not choose the same priority for multiple libraries.
- You can select your order of preference for endogenous library alignment.
- Advanced Endogenous Alignment Options (initially minimized)
- Maximum Number of Endogenous Mismatches Allowed - This option will alter the maximum number of mismatches allowed during endogenous alignment. Range from 0 to 3. Default of 1.
- Minimum Fraction of Read Remaining After Soft Clipping - This option will alter the minimum fraction of the read that must remain following soft-clipping (in a local alignment). Default of 0.9.
- Downsample RNA Reads for Transcriptome Alignments - This option will allow you to downsample your RNA reads after assigning reads to the various transcriptome libraries.
This may be useful for normalizing very different yields. If you want to downsample, click the checkbox and then put the number of RNA reads to which you want to downsample.
We recommend a minimum of 100000, which is the default if you choose to downsample.
- Exogenous Alignment Options
- You can select your preference for exogenous library alignment. There are three options:
- Endogenous-only - Disables mapping to exogenous miRNAs.
- Endogenous + exogenous (miRNA) - Maps to exogenous miRNAs in miRBase (i.e., from plants and viruses).
- Endogenous + exogenous (miRNA + Genome) - Maps to exogenous miRNAs in miRBase AND the genomes of all sequenced species in Ensembl/NCBI.
- Note that if you choose either the second or third option, then you cannot turn off any of the endogenous mappings above.
- If you have already turned off any endogenous mappings, then you cannot choose either the second or third option.
- You can select your preference for exogenous library alignment. There are three options:
- Advanced Exogenous Alignment Options (initially minimized)
- Maximum Number of Exogenous Mismatches Allowed - This option will alter the maximum number of mismatches allowed during exogenous alignment. Range from 0 to 1. Default of 0.
- Other Advanced Options (initially minimized)
- Remote Storage Area - This option will allow you to choose a remote storage (FTP) area where your result files will be uploaded.
These result files will then be accessible via FTP client. You can learn more by visiting the Using Remote (FTP) Storage for exceRpt help page. - Suppress Individual Sample Emails - This option will turn off the individual runExceRpt emails for each sample as it gets processed.
If your submission includes dozens of samples and you'd prefer to not receive dozens of emails, click the checkbox. - Upload Full Results - This option will upload all of the result files for each sample, as opposed to the core, most important files that are normally uploaded.
For example, a full results archive will contain all of the full alignment .bam files generated by the pipeline.
This archive will be much larger than the core results archive and will significantly eat into your allotted storage space, so only select this option if necessary!
To learn more about the storage quotas we have recently implemented, view the Understanding Your Storage Options with exceRpt page.
- Remote Storage Area - This option will allow you to choose a remote storage (FTP) area where your result files will be uploaded.
Example Data for Running exceRpt Small RNA-seq Pipeline¶
In this example, we have used four samples from deep sequencing experiments of barcoded small RNA cDNA libraries to profile microRNAs in
cell-free serum and plasma from human volunteers. These samples were analyzed using the exceRpt Small RNA-seq Pipeline.
The sample input FASTQ files and output results and plots can be found here:

- Under the group
Examples and Test Data, select the DatabaseexceRpt small RNA-seq Pipeline - Example Data. - The compressed input files can be found in the archive:
Files » placental_serum_plasma_SRA_Study_SRP018255_4_samples.tar.gz.- These same compressed input files can be found unarchived in the following folder:
Files » Placental smallRNAs SRA SRP018255 4-samples.
- These same compressed input files can be found unarchived in the following folder:
- Pipeline results can be found under the
Files » exceRptPipeline_v4.6.2 » Circulating microRNAs from serum plasma - Study SRP18255folder.- Each sample has its own dedicated folder (
sample_C1_non_pregnant1_SRR822433_fastq,sample_C3_non_pregnant3_SRR822434_fastq, etc.).
- Each sample has its own dedicated folder (
- Core result files for a given sample can found in its
CORE_RESULTSfolder. - Post-processing plots and results for all samples can be found in the
postProcessedResults_v4.6.3folder (under theCirculating microRNAs from serum plasma - Study SRP18255folder).
exceRpt Small RNA-seq Pipeline Workflow (Implemented in the Genboree Workbench)¶
The exceRpt Small RNA-seq Pipeline is for the processing and analysis of RNA-seq data generated to profile small-exRNAs.
The pipeline is highly modular, allowing the user to define the libraries containing smallRNA sequences that are used
during RNA-seq read-mapping, including an option to provide a library of spike-in sequences to allow absolute quantitation
of small-RNA molecules. It also performs automatic detection and removal of 3' adapter sequences.
The output data includes abundance estimates for each of the requested libraries, a variety of quality control metrics
such as read-length distribution, summaries of reads mapped to each library, and detailed mapping information for each read mapped to each library.
exRNA Data Analysis Results¶
To better understand the results generated by exceRpt, check out the exRNA Data Analysis page.
Finally, after the pipeline finishes processing all submitted samples, a separate post-processing tool (processPipelineRuns)
is run on all successful pipeline outputs. This tool generates useful summary plots and tables that can be used to compare
and contrast different samples.
Currently Supported Genomes¶
- Human Genome version - hg38
- Human Genome version - hg19
- Mouse Genome version - mm10
Sources of smallRNA Libraries¶
- rRNAs from 45S, 5S, and mt_rRNA sequences for human and mouse
- miRNAs from miRBase version 21
- tRNAs from gtRNAdb
- piRNAs from piRNABank (removed duplicate sequences)
- Annotations from Gencode version 24 (hg38), version 18 (hg19), version M9 (mm10)
- CircularRNAs from circBase
Workflow¶
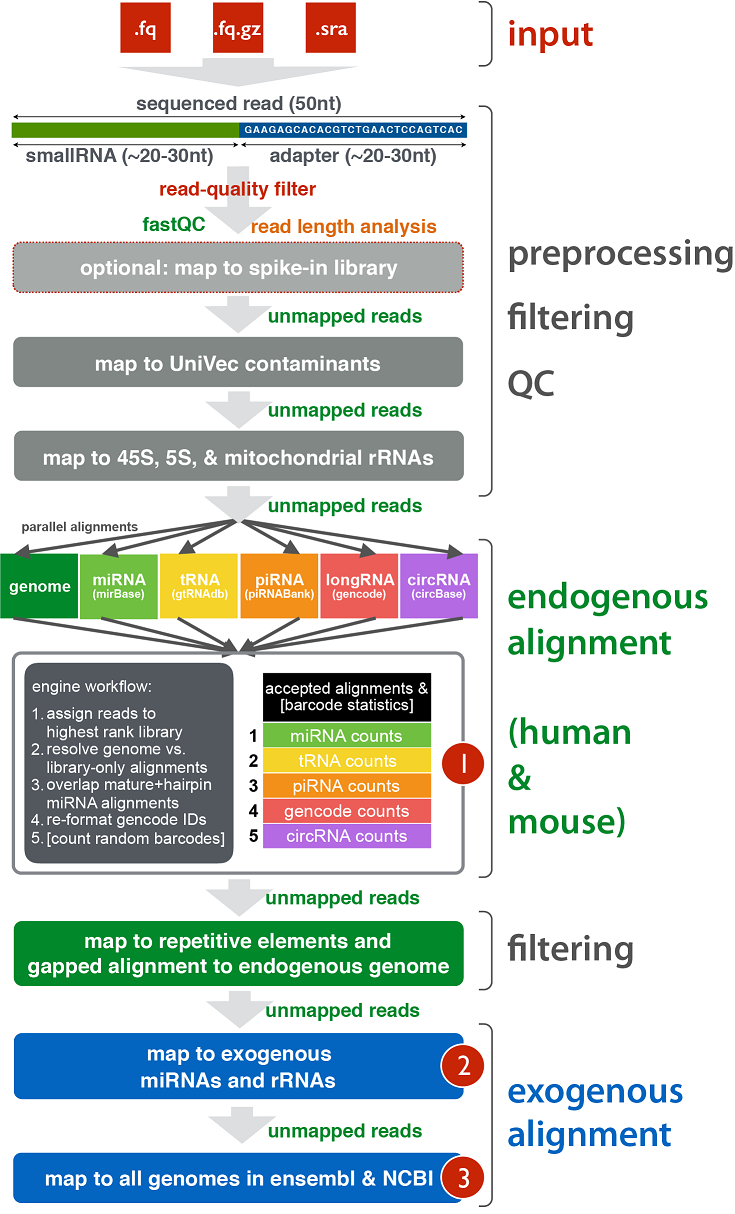
Example of Core Results from Workflow¶

Post-processing of Samples¶
After all samples have been processed through this pipeline, the Generate Summary Report for exceRpt Results tool will take
successful samples and perform post-processing on them.
This post-processing step will generate useful plots and tables that will allow you to compare and contrast samples.
A description of all generated files can be found in the table below (partially taken from the exceRpt GitHub page):
| File Name | Description of File |
| QC Data | |
| exceRpt_DiagnosticPlots.pdf | All diagnostic plots automatically generated by the tool |
| exceRpt_readMappingSummary.txt | Read-alignment summary including total counts for each library |
| exceRpt_ReadLengths.txt | Read-lengths (after 3' adapters/barcodes are removed) |
| Raw Transcriptome Quantifications | |
| exceRpt_miRNA_ReadCounts.txt | miRNA read-counts quantifications |
| exceRpt_tRNA_ReadCounts.txt | tRNA read-counts quantifications |
| exceRpt_piRNA_ReadCounts.txt | piRNA read-counts quantifications |
| exceRpt_gencode_ReadCounts.txt | gencode read-counts quantifications |
| exceRpt_circularRNA_ReadCounts.txt | circularRNA read-counts quantifications |
| exceRpt_biotypeCounts.txt | read-counts quantified for different biotypes |
| Normalized Transcriptome Quantifications | |
| exceRpt_miRNA_ReadsPerMillion.txt | miRNA RPM quantifications |
| exceRpt_tRNA_ReadsPerMillion.txt | tRNA RPM quantifications |
| exceRpt_piRNA_ReadsPerMillion.txt | piRNA RPM quantifications |
| exceRpt_gencode_ReadsPerMillion.txt | gencode RPM quantifications |
| exceRpt_circularRNA_ReadsPerMillion.txt | circularRNA RPM quantifications |
| Exogenous Output | |
| exceRpt_exogenousGenomes_TaxonomyTrees_aggregateSamples.pdf | aggregate taxonomy tree for exogenous genomes |
| exceRpt_exogenousGenomes_TaxonomyTrees_perSample.pdf | per-sample taxonomy trees for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomyCumulative_ReadCounts.txt | descendant read counts for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomyCumulative_ReadsPerMillion.txt | descendant RPM counts for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomySpecific_ReadCounts.txt | direct read counts for exogenous genomes |
| exceRpt_exogenousGenomes_taxonomySpecific_ReadsPerMillion.txt | direct RPM counts for exogenous genomes |
| exceRpt_exogenousRibosomal_TaxonomyTrees_aggregateSamples.pdf | aggregate taxonomy tree for exogenous rRNAs |
| exceRpt_exogenousRibosomal_TaxonomyTrees_perSample.pdf | per-sample taxonomy trees for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomyCumulative_ReadCounts.txt | descendant read counts for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomyCumulative_ReadsPerMillion.txt | descendant RPM counts for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomySpecific_ReadCounts.txt | direct read counts for exogenous rRNAs |
| exceRpt_exogenousRibosomal_taxonomySpecific_ReadsPerMillion.txt | direct RPM counts for exogenous rRNAs |
| R Objects | |
| exceRpt_smallRNAQuants_ReadCounts.RData | All raw data (binary R object) |
| exceRpt_smallRNAQuants_ReadsPerMillion.RData | All normalized data (binary R object) |
| Misc. | |
| exceRpt_adapterSequences.txt | 3' adapter sequences associated with each sample |
| exceRpt_sampleGroupDefinitions.txt | sample group associated with each sample (not used in Genboree) |
Explanation of Exogenous Output¶
The taxonomy specific read counts file contains the number of reads that map directly to each node in the sample's taxonomy tree.
The taxonomy cumulative read counts file contains the number of reads that map to the descendants of each node in the sample's taxonomy tree.
Importantly, the reads counted for a given node in the taxonomy specific read counts file are NOT counted as part of the cumulative counts in the other file
(all of the node's descendants are counted, but not the node itself).
- 1 superkingdom 10239 1 Viruses 131 182 141 15 122 136 97 100 115
- 1 superkingdom 10239 1 Viruses 0 0 0 0 0 0 0 0 0
This means that no reads mapped directly to the Viruses node, but a considerable number of reads mapped to descendants of Viruses.
Similarly, say we have this line in the cumulative file:- 1 no rank 131567 1 cellular organisms 649526 737020 724361 200600 316643 412608 365094 374525 473008
- 1 no rank 131567 1 cellular organisms 791672 806489 734921 211454 320147 398621 348969 407637 494698
This means that a considerable number of reads mapped both directly to the cellular organisms node as well as descendants of that node.
In other words, if you want to get a full count of all of the reads that aligned to a given node and its descendants,
you need to ADD TOGETHER the numbers in both the taxonomy cumulative and taxonomy specific files for that node.
You can also find plots of the exogenous trees in the .pdf plot files.
First, the TaxonomyTrees_perSample.pdf file contains a plot for each sample.
The percentage within each node is the ratio of the node's summed cumulative + specific reads to the root node's summed cumulative + specific reads.
Second, the TaxonomyTrees_aggregateSamples.pdf file contains a single plot that condenses all samples into a single, averaged tree.
The percentage within each node is the ratio of that node's summed cumulative + specific reads averaged across all samples to the root node's summed cumulative + specific reads averaged across all samples.
Example of Post-processed Result Files¶
Below, we can see what the post-processing files look like on the Genboree Workbench (found in the Examples and Test Data Group):

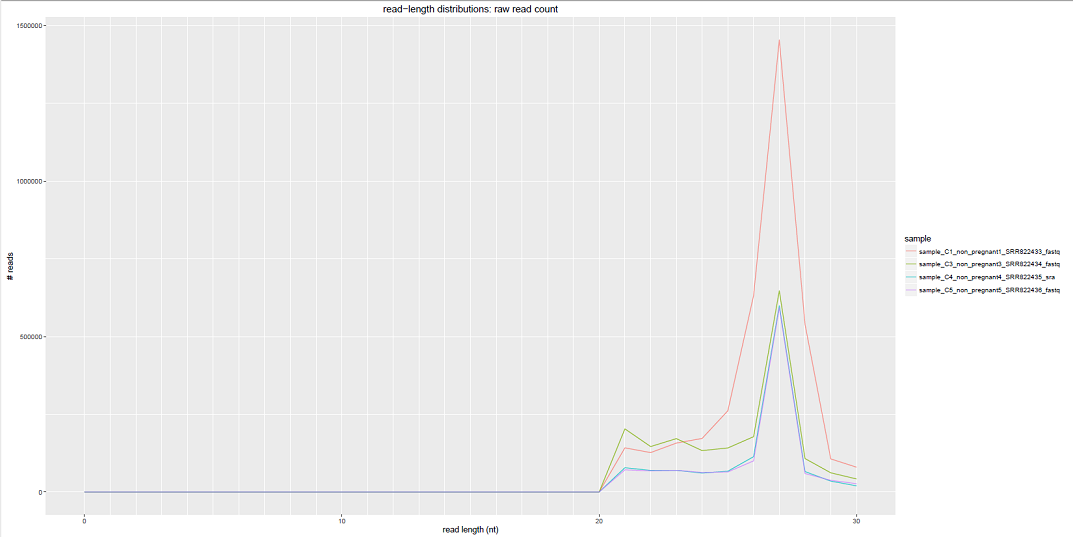
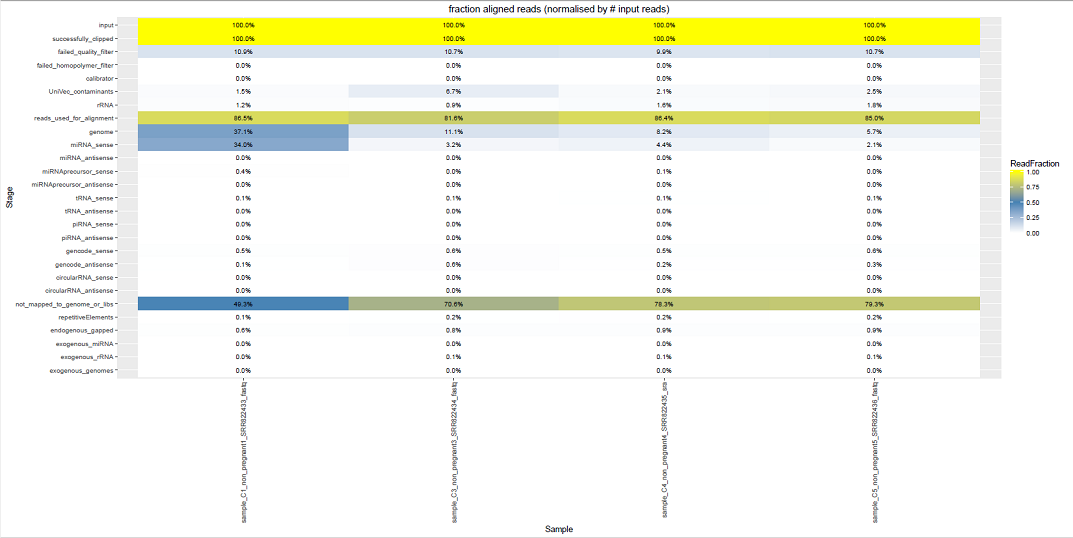
Comparative Plots¶
We can see some examples below of the comparative plots available in the DiagnosticPlots.pdf generated by the post-processing tool.
Each plot contains data from four different samples (the example data).


Bioinformatics Tools Used in This Pipeline (4th Gen)¶
- FASTX-Toolkit v0.0.14
- Bowtie2 v2.2.6
- Samtools v1.3.1
- SRA-Toolkit v2.3
- R v3.2
- FastQC v0.11.2
- STAR v2.4.2a
References and Attributions¶
- This tool has been developed by the Data Integration and Analysis Component (DIAC) of the Extracellular RNA Communication Consortium
- exceRpt small RNA-seq pipeline was developed by Robert Kitchen at the Gerstein Lab at Yale University
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21. doi:10.1093/bioinformatics/bts635. [Pubmed]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. NAR 2011 39 (Database Issue):D152-D157 [Pubmed]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012 Mar 4; 9 : 357-359. [PubMed]
- Integrated into the Genboree Workbench by Sai Lakshmi Subramanian and William Thistlethwaite
at the Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX.
This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact "exRNA Team":brl-exrna@bcm.edu with questions or comments, or for help using it on your own data.