Overview
- Fold Change Calculation Using DESeq2
- Introduction
- Preliminary Steps for Setting Up Any Analysis in the Genboree Workbench
- Create a Group for Your Analysis
- Create a Database for Your Analysis
- Upload your Data File(s)
- Step-by-step Instructions to Set Up Job
- Example Data for Running DESeq2
- Output Files Generated by Job
- References and Attributions
Fold Change Calculation Using DESeq2¶
Introduction¶
This tool will test samples for differential expression using DESeq2 (version 1.6.3).
Currently, the tool allows you to test a given factor (disease, for example) across two different factor levels (control versus Alzheimer's disease, for example).
We will continue to develop this tool and will add new features (like allowing analysis over multiple factors) in the coming months.
Preliminary Steps for Setting Up Any Analysis in the Genboree Workbench¶
Create a Group for Your Analysis¶

What is a  Group? Group? |
A "Group" contains Databases and Projects and controls access to all content within. You control access to your Group(s), and who is a member of your group. You can also belong to multiple Groups (i.e. collaborators). |
This step is optional. You can also use your default/existing group.
Create a Database for Your Analysis¶
What is a  Database? Database? |
A "Database" contains Tracks, Lists, Sample Sets, Samples, and Files. Each database can be associated with a reference genome. |
This step is required.
Make sure that you pick the proper reference sequence genome (hg19, for example) when creating your database.
Note that we do not have an entry for hg38 or mm10 in our "Reference Sequence" list.
If your data is associated with either of these reference sequence genomes, follow the directions below.
Create a Database for hg38 or mm10
In order to create a database associated with hg38 or mm10, select the "User Will Upload" option for "Reference Sequence"
and provide appropriate values for the Species and Version text boxes as given below:
| Your Genome of Interest | Species | Version |
| Human genome hg38 | Homo sapiens | hg38 |
| Mouse genome mm10 | Mus musculus | mm10 |
If your genome of interest is not available, please contact the exRNA Team for help.
Upload your Data File(s)¶
What types of files can be uploaded?
The Fold Change Calculating Using DESeq2 tool accepts exactly two text files as input.
One file should contain your miRNA read counts, with rows corresponding to miRNA identifiers and columns corresponding to individual sample names.
The other file should contain your sample descriptors, with rows corresponding to individual sample names and columns corresponding to factor names ("condition", "biofluid", etc.).
Step-by-step Instructions to Set Up Job¶
- Drag exactly two text files (with the formatting described above) into the
Input Datapanel. You can also drag a folder or file entity list if it contains both text files. - Drag a
Databaseto theOutput Targetspanel to store results. - Select
Transcriptome»Differential Expression Analysis»Fold Change Calculating Using DESeq2from the Toolset menu.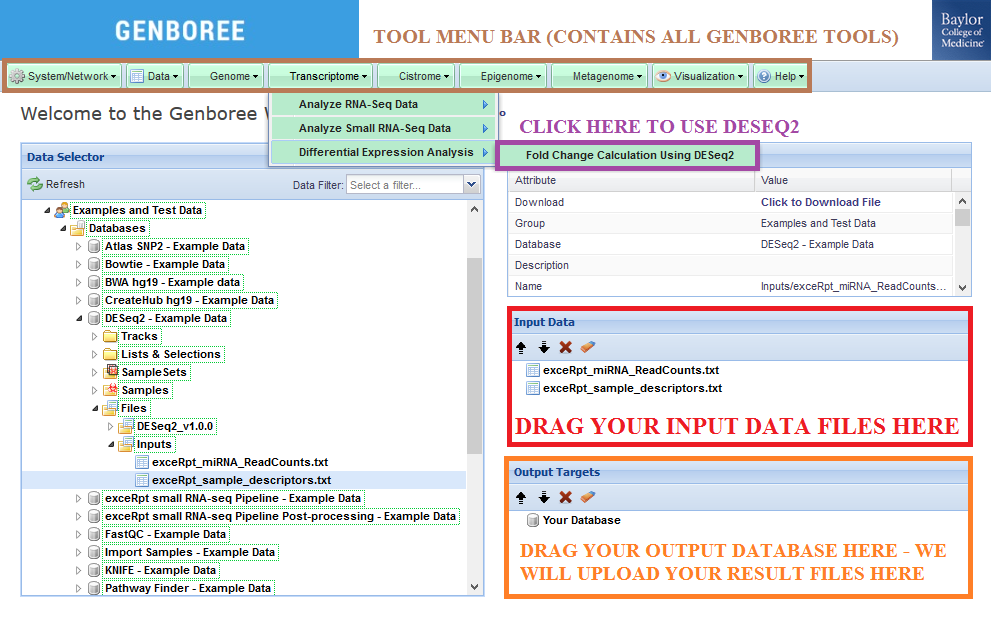
- Fill in the analysis name for your tool job. We recommend keeping a timestamp in your analysis name!
- Fill in the factor name and the corresponding factor levels for your analysis. For example, if I was using the tutorial files and wanted to examine the "disease" factor and compare "AD" (Alzheimer's disease) to "CONTROL" (healthy controls), I would put the following values:
- Factor Name: disease
- Factor Level 1: AD
- Factor Level 2: CONTROL
- Select the different ERCC-related submission settings if you are a member of the ERCC. If you are not, then ignore this section.
- Choose to upload your results to a remote storage area if you wish to do so. More information about this option can be found here.
- Submit your job. Upon completion of your job, you will receive an email.
- Download the results of your analysis from your
Database. The results data will end up under theDESeq_v1.0.0folder in theFilesarea of your output database.- Within that folder, your
Analysis Namewill be used as a sub-folder to hold the files generated by that run of the tool. - Open this sub-folder to see your results.
- Select any of the output files (explained in more detail below) and then click the link
Click to Download Filefrom theDetailspanel to download that output file.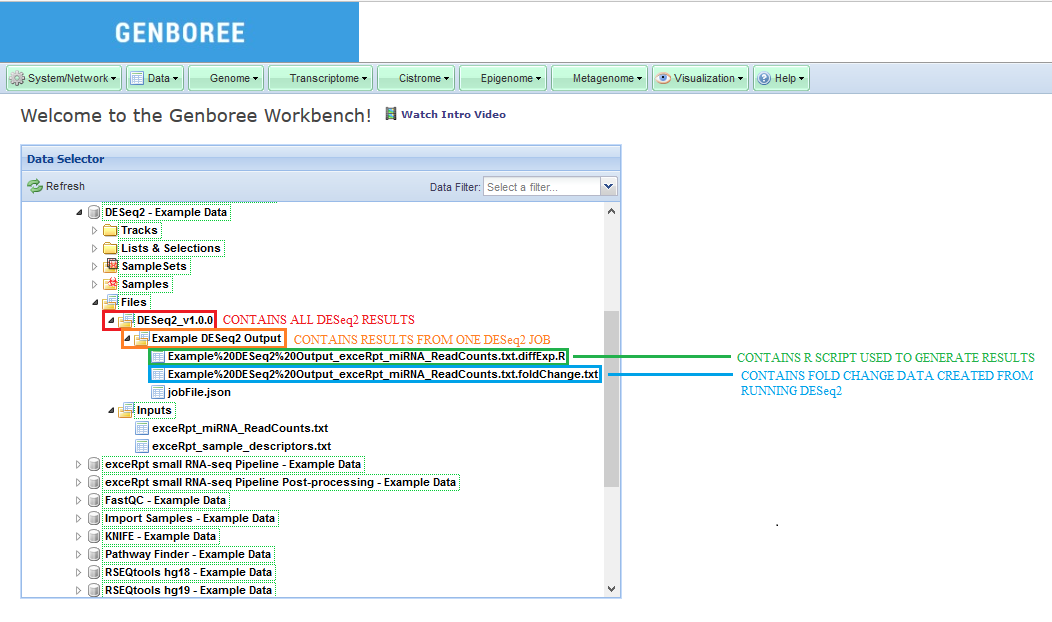
- Within that folder, your
Example Data for Running DESeq2¶
In this example, we have used a set of miRNA read counts processed by exceRpt for 181 different samples (found in exceRpt_miRNA_ReadCounts.txt).
We have also used a sample descriptor document which contains information about disease and biofluid for each of the 181 samples (found in exceRpt_sample_descriptors.txt).
The sample input files and output results can be found here:
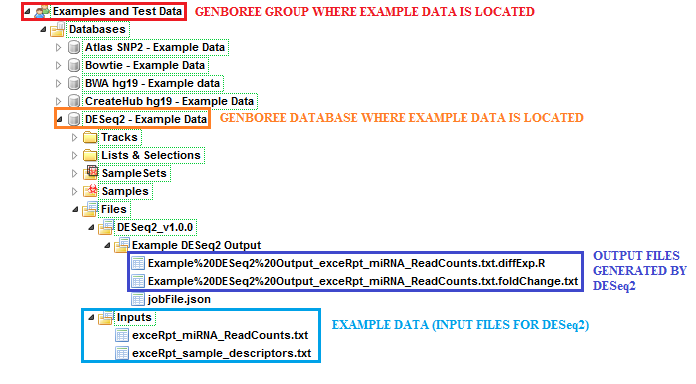
- Under the group
Examples and Test Data, select the databaseDESeq2 - Example Data. - Both input files can be found in the folder:
Files » Inputs. - DESeq2 results can be found under the
Files » DESeq2_v1.0.0 » Example DESeq2 Outputfolder in this database.
Output Files Generated by Job¶
After your job successfully completes, you will be able to download 2 different output files:
- A _foldChange.txt file that contains the results from your DESeq2 analysis.
- A _diffExp.R file that is the R script used to generate your results.
References and Attributions¶
- M. I. Love, W. Huber, S. Anders: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014, 15:550. http://dx.doi.org/10.1186/s13059-014-0550-8
- Integrated into the Genboree Workbench by William Thistlethwaite and Sai Lakshmi Subramanian at the Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX.
This tool has been deployed in the context of the exRNA Communication Consortium (ERCC).
Please contact the exRNA Team with questions or comments, or for help using it on your own data.