How do I prepare, upload, and evaluate 27k / 450k data? (Multi-block data format)
Introduction¶
27k and 450k References¶
In order to grasp the general procedure of understanding and utilizing the 27k and 450k output we recommend some of the following manuscripts:- 27k
- DNA methylation profiling reveals a predominant immune component in breast cancers
- 450k
- Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome
- Evaluation of the Infinium Methylation 450K technology
Tutorial Data Set¶
In order to illustrate how to use the Genboree Workbench to evaluate 27k / 450k data, we're going to demonstrate how to utilize a publicly available data set:- Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome
- http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE29290
- Download and decompress this file:
- http://www.ncbi.nlm.nih.gov/geosuppl/?acc=GSE29290&file=GSE29290%5FMatrix%5FProcessed%2Etxt%2Egz
- GSE29290_Matrix_Processed.txt.gz
- GSE29290_Matrix_Processed.txt.zip
- GSE29290_Matrix_Processed.txt.gz
- http://www.ncbi.nlm.nih.gov/geosuppl/?acc=GSE29290&file=GSE29290%5FMatrix%5FProcessed%2Etxt%2Egz
- Download and decompress this file:
- The data can be tab or comma delimited format
- One column has to represent the probe ID
- I.e. cg00000029
- One column has to represent the probe score for each sample (i.e. <sample>.AVG_Beta, <sample>.M-value, etc.)
ID_REF Sample_1.AVG_Beta Sample_1.Detection Pval Sample_2.AVG_Beta Sample_2.Detection Pval Sample_3.AVG_Beta Sample_3.Detection Pval cg00000029 0.8296142 0 0.852155 0 0.8956234 0 ... cg00000108 0.8492596 0 0.8898684 0 0.9276204 0 ... cg00000109 0.8247395 0 0.8609225 0 0.8725377 0 ... cg00000165 0.8228635 0 0.8665444 0 0.8800115 0 ... ... ... ... ... ... ... ... ...
Preparing Processed Matrix for Genboree Workbench¶
We need to take the processed matrix and prepare it for import within the 'Array Data Importer' utility. Please read the help on this tool, but we will also post the necessary file format here:
Format:
##trackName=myTrackName_1:trackSubName_1 #Probe_Name<tab>Score_Value <ProbeID_1><tab><ProbeScore_1> <ProbeID_2><tab><ProbeScore_2> <ProbeID_3><tab><ProbeScore_3> ... ##trackName=myTrackName_2:trackSubName_2 #Probe_Name<tab>Score_Value <ProbeID_1><tab><ProbeScore_1> <ProbeID_2><tab><ProbeScore_2> <ProbeID_3><tab><ProbeScore_3> ...
Actual implementation of above sample (in data format):
| ##trackName=Sample_1.AVG_Beta:Subset_avg_beta | |
| #Probe | Score |
| cg00000029 | 0.8296142 |
| cg00000108 | 0.8492596 |
| cg00000109 | 0.8247395 |
| cg00000165 | 0.8228635 |
| ##trackName=Sample_2.AVG_Beta:Subset_avg_beta | |
| #Probe | Score |
| cg00000029 | 0.852155 |
| cg00000108 | 0.8898684 |
| cg00000109 | 0.8609225 |
| cg00000165 | 0.8665444 |
- A header line for each separate sample followed by the column headers
- This will repeat every time you wish to import an additional sample
- A unique track name
- Probe IDs that exist within the ROI (region of interest) annotation track
- Numerical values for the Score data
- GSE29290_Matrix_Processed.txt-array_format-450k.tsv.zip
Preparing Metadata for 27k & 450k Data Sets¶
In order to be able to utilize the Genboree Workbench to analyze your array data, it is most convenient if you produce some metadata for your samples. Providing metadata for your samples will allow you to more easily create sets of tracks (called Track Entity Lists) in order to be able to evaluate your samples in a variety of groups.
Creating Track Metadata- This example has 2 metadata columns
- cell_type
- Colorectal_cancer
- Colorectal_cancer_knock_out
- Breast_normal
- Breast_tumor
- experiment_type
- 450k
- cell_type
| #name | cell_type | experiment_type |
| Sample_1.AVG_Beta:450k_avg_beta | Colorectal_cancer | 450k |
| Sample_2.AVG_Beta:450k_avg_beta | Colorectal_cancer | 450k |
| Sample_3.AVG_Beta:450k_avg_beta | Colorectal_cancer | 450k |
| Sample_4.AVG_Beta:450k_avg_beta | Colorectal_cancer_knock_out | 450k |
| Sample_5.AVG_Beta:450k_avg_beta | Colorectal_cancer_knock_out | 450k |
| Sample_6.AVG_Beta:450k_avg_beta | Colorectal_cancer_knock_out | 450k |
| Sample_7.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_8.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_9.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_10.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_11.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_12.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_13.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_14.AVG_Beta:450k_avg_beta | Breast_normal | 450k |
| Sample_15.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
| Sample_16.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
| Sample_17.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
| Sample_18.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
| Sample_19.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
| Sample_20.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
| Sample_21.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
| Sample_22.AVG_Beta:450k_avg_beta | Breast_tumor | 450k |
- GSE29290_full_track_metadata.tsv
Using the Genboree Workbench to Evaluate 27k & 450k Data Sets - Step by Step¶
- Create a new Database
- Drag your group into the 'Output Targets' window
- Click 'Data' -> 'Databases' -> 'Create Database'
- Select 'Template: Human (hg19)
- Enter a Database Name
- Click Submit
- Create a new Project
- Drag your group into the 'Output Targets' window
- Click 'Data' -> 'Projects' -> 'Create Project'
- Enter Project Name
- Click Submit
- Upload your prepared array data ('GSE29290_Matrix_Processed.txt-array_format-450k.tsv.zip')
- (Remove your Group from the 'Output Targets' window
- Drag your Database into the 'Output Targets' window
- Click 'Data' -> 'Files' -> 'Transfer File'
- Choose your file
- Click Submit
- Import your array data
- Drag your Database into the 'Output Targets' window
- Drag your file into the 'Input Data' window
- This file is located in your_group -> Databases -> your_database -> Files
- Click 'Data' -> 'Tracks' -> 'Import' -> 'Array Data'
- Select 'Hs Methylation:450k'
- You would select 'Hs Methylation:27k' if you are using 27k data
- Select File Format 'Muti-block'
- Click Submit
- Wait for success email
Hello Kevin Riehle, Your Array Data Importer job has completed successfully. JOB SUMMARY: JobID : wbJob-arraydataimporter-1347059333_775361 The following array/probe file has been imported: GSE29290_Matrix_Processed.txt-array_format-450k.tsv The following tracks were uploaded in the target database: Sample_19.AVG_Beta:450k_avg_beta Sample_2.AVG_Beta:450k_avg_beta Sample_1.AVG_Beta:450k_avg_beta Sample_3.AVG_Beta:450k_avg_beta Sample_15.AVG_Beta:450k_avg_beta Sample_17.AVG_Beta:450k_avg_beta Sample_16.AVG_Beta:450k_avg_beta Sample_14.AVG_Beta:450k_avg_beta Sample_12.AVG_Beta:450k_avg_beta Sample_8.AVG_Beta:450k_avg_beta Sample_9.AVG_Beta:450k_avg_beta Sample_7.AVG_Beta:450k_avg_beta Sample_13.AVG_Beta:450k_avg_beta Sample_21.AVG_Beta:450k_avg_beta Sample_22.AVG_Beta:450k_avg_beta Sample_5.AVG_Beta:450k_avg_beta Sample_11.AVG_Beta:450k_avg_beta Sample_6.AVG_Beta:450k_avg_beta Sample_10.AVG_Beta:450k_avg_beta Sample_4.AVG_Beta:450k_avg_beta Sample_18.AVG_Beta:450k_avg_beta Sample_20.AVG_Beta:450k_avg_beta The Genboree Team ...Add Track Metadata
- Upload track metadata file
- GSE29290_track_metadata.tsv
- Drag your File (i.e. 'GSE29290_full_track_metadata.tsv') to 'Input Data' window
- Drag your Database to 'Output Targets' window
- Click 'Data' -> 'Tracks' -> 'Import' -> 'Track Metadata'
- Uncheck 'Create New Tracks?'
- Click Submit
- Drag your Database into 'Input Data'
- Click 'Visualization' -> 'View Track Grid'
- X-axis attribute
- cell_type
- Y-axis attribute
- experiment_type
- Click Submit
- Click the blue hyperlink 'Launch Grid Viewer'
- X-axis attribute
- Select the (8) Breast_normal cell, the (8) Breast_tumor cell, the (3) Colorectal_cancer cell, and the (3) Colorectal_cancer_knock_out cell.
- Click 'Selections' -> 'Save Selections'
- Select your Group
- Select your Database
- Type in a name (i.e. 'all22samplesTrackEntityList_A')
- Click 'Save Selections'
- Select the (8) Breast_tumor cell
- Click 'Selections' -> 'Save Selections'
- Select your Group
- Select your Database
- Type in a name (i.e. ''all22samplesTrackEntityList_B')
- Click 'Save Selections'
- Select the (8) Breast_normal cell
- Click 'Selections' -> 'Save Selections'
- Select your Group
- Select your Database
- Type in a name (i.e. 'Breast_normal_450k')
- Click 'Save Selections'
- Select the (8) Breast_tumor cell
- Click 'Selections' -> 'Save Selections'
- Select your Group
- Select your Database
- Type in a name (i.e. 'Breast_tumor_450k')
- Click 'Save Selections'
Heatmap¶
- Run Heatmap on All Tracks
- (Clear any entries in the 'Input Data' window if they exist)
- Drag your two track entity lists into the 'Input Data' window (i.e. 22samplesTrackEntityList_A and 22samplesTrackEntityList_B)
- These track entity lists are located in your_group -> Databases -> your_database -> Lists & Selections -> List of Tracks
- Drag your desired ROI (regions of interest) track into the 'Input Data' window
- For example, you can use the Promoters:LCP ROI track
- This track is located in ROI Repository -> Databases -> ROI Repository - hg19 -> Tracks -> Class: Regulation
- For example, you can use the Promoters:LCP ROI track
- Drag your Database into the 'Output Targets' window
- Drag your Project into the 'Output Targets' window
- Click 'Epigenome' -> 'Compute Similarity Matrix (heatmap)'
- Click Submit
- Wait for a confirmation email
Hello Kevin Riehle,
Your Epigenomic Experiment Heatmap Tool job completed successfully.
Job Summary:
JobID - wbJob-epigenomicsheatmap-1347635780_213632
Analysis Name - EpigenomeExpHeatmap2012-09-14-10:15:54
Inputs:
1. Entitylist - 450k_1
2. Entitylist - 450k_2
3. Trk - Promoters%3ALCP
Outputs:
1. Db - 450ktest_full
2. Prj - 450k
Settings:
analysisName - EpigenomeExpHeatmap2012-09-14-10:15:54
clusterQueue - gbMultiCore
color - Spectral
dendograms - both
density - histogram
distfun - dist
fixedResolution - medium
hclustfun - hclust
height - 8
key - TRUE
keySize - 0.75
quantileNormalized - true
removeNoDataRegions - true
spanAggFunction - avg
trace - none
width - 10
- The Genboree Team
Result File Location in the Genboree Workbench:
http://www.genboree.org/java-bin/project.jsp?projectName=450k
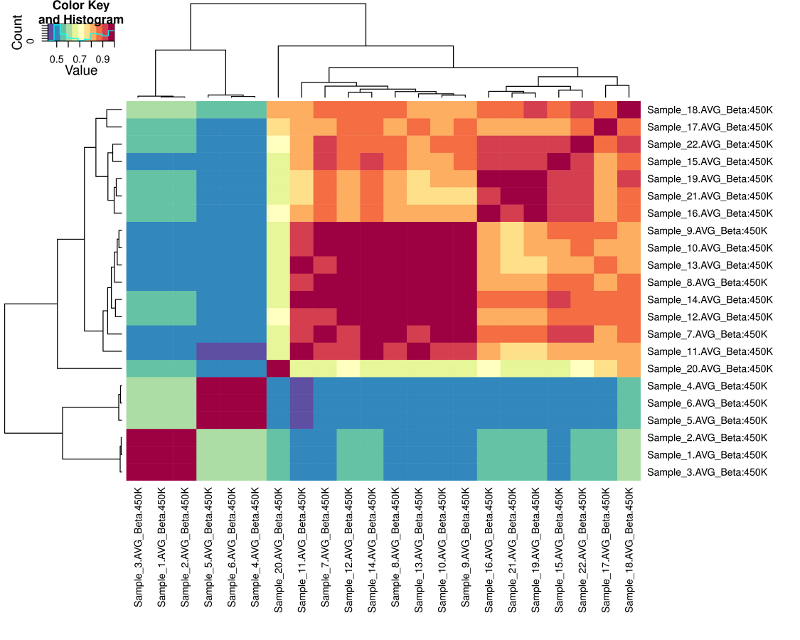
- You will see that we witness clustering among:
- 8 Breast_normal
- 7 Breast_tumor (Sample_20 is an outlier)
- 3 Colorectal_cancer
- 3 Colorectal_cancer_knock_out
LIMMA¶
Run LIMMA- Drag your first track entity list into the 'Input Data' window (i.e. 'Breast_normal_450k')
- Drag your second track entity list into the 'Input Data' window (i.e. 'Breast_tumor_450k')
- Drag your ROI (regions of interest) track into the 'Input Data' window (i.e. 'Promoters:ALL)
- This track is located in ROI Repository -> Databases -> ROI Repository - hg19 -> Tracks -> Class: Regulation
- Drag your Database into the 'Output Targets' window
- Drag your Project into the 'Output Targets' window
- Click 'Epigenome' -> 'Analyze Signals' -> 'Compare by LIMMA' -> 'Tracks'
- Click Submit
- Wait for confirmation emails:
- "Genboree: Your Epigenomic Experiment Sets Comparison Using Limma job is complete"
- "LFF API Upload [SUCCESS]"
SPARK¶
Run SPARK- Drag your first track entity list into the 'Input Data' window (i.e. 'Breast_normal_450k')
- Drag your second track entity list into the 'Input Data' window (i.e. 'Breast_tumor_450k')
- Drag your ROI (regions of interest) track into the 'Input Data' window (i.e. 'Promoters:ALL)
- This track is located in ROI Repository -> Databases -> ROI Repository - hg19 -> Tracks -> Class: Regulation
- Drag your Database into the 'Output Targets' window
- Click 'Epigenome' -> 'Analyze Signals' -> 'Cluster by Spark'
- Select your ROI Track
- Single click on your ROI track (i.e. Promoters:LCP)
- Customize the settings or leave the defaults
- Optionally change track colors
- I.e. change samples 15-22 to 'green' for Data Track Colors
- Click Submit
- Wait for confirmation email and follow directions
- Select your ROI Track
{kind=link}
Also available in: PDF